WO1996041865A1 - Rapamcycin-based regulation of biological events - Google Patents
Rapamcycin-based regulation of biological events Download PDFInfo
- Publication number
- WO1996041865A1 WO1996041865A1 PCT/US1996/009948 US9609948W WO9641865A1 WO 1996041865 A1 WO1996041865 A1 WO 1996041865A1 US 9609948 W US9609948 W US 9609948W WO 9641865 A1 WO9641865 A1 WO 9641865A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- domain
- rapamycin
- cells
- fkbp
- frb
- Prior art date
Links
- 0 C[C@](CC(CC1)CC(*)[C@@]1O)[C@](CC([C@](C)C=C(C)C(*)[C@@](*)C(C(C)C[C@](C)C=CCCC=C(C)[C@](C[C@](CCC1C)OC1(C(C(C1C2CCCC*1)=O)=C1CC1)O)OC)=O)=O)OC2=O Chemical compound C[C@](CC(CC1)CC(*)[C@@]1O)[C@](CC([C@](C)C=C(C)C(*)[C@@](*)C(C(C)C[C@](C)C=CCCC=C(C)[C@](C[C@](CCC1C)OC1(C(C(C1C2CCCC*1)=O)=C1CC1)O)OC)=O)=O)OC2=O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/10—Cells modified by introduction of foreign genetic material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
- C12N9/12—Transferases (2.) transferring phosphorus containing groups, e.g. kinases (2.7)
- C12N9/1205—Phosphotransferases with an alcohol group as acceptor (2.7.1), e.g. protein kinases
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/37—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from fungi
- C07K14/39—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from fungi from yeasts
- C07K14/395—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from fungi from yeasts from Saccharomyces
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/715—Receptors; Cell surface antigens; Cell surface determinants for cytokines; for lymphokines; for interferons
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1055—Protein x Protein interaction, e.g. two hybrid selection
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/90—Isomerases (5.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2840/00—Vectors comprising a special translation-regulating system
- C12N2840/20—Vectors comprising a special translation-regulating system translation of more than one cistron
- C12N2840/203—Vectors comprising a special translation-regulating system translation of more than one cistron having an IRES
Definitions
- Rapamycin (I) is a natural product which binds to a FK506-binding protein, FKBP, with high affinity to form a rapamycin.FKBP complex. Reported Kd values for that interaction are as low as 200 pM.
- the rapamycin.FKBP complex binds with high affinity to the large cellular protein FRAP to form a tripartite, [FKBP:rapamycin]:[FRAP]. complex.
- rapamycin acts as a dimerizer or adapter to join FKBP to FRAP.
- FKBP:rapamycin complex is referred to as the FRB domain, which is discussed in detail below.
- FKBP12 rapamycin-dependent association of FKBP12 and a large mammalian protein termed FRAP, RAFT1 or RAPT1 and its yeast homologs DRR and TOR have been described by several research groups. See e.g., Brown et al, 1994, Nature 369:756-758; Sabatini et al, 1994, Cell 78:35-43; Chiu et al, 1994, Proc. Natl. Acad. Sci. USA
- FKBPs FK506 binding proteins
- rapamycin While rapamycin, like FK506, is a natural dimerizer of proteins, and is capable of dimerizing appropriately designed chiemric proteins, its significant biological activities, including potent immunosuppressive activity, rather severely limit its use in engineered biological switches, particularly for use in animals.
- This invention harnesses the dimerizing potential of rapamycin (and related compounds) while avoiding its profound, inherent limitations.
- This invention concerns new configurations for biological switches and provides new methods and materials for regulating biological events, particularly in animal cells.
- Those biological events include, for example, gene transcription, activation of an intracellular signal transduction pathway leading for example to gene expression or apoptotic cell death, gene knock-out, blockade of expression of a gene, and inhibition of the function of a gene product.
- the invention relies upon two types of chimeric proteins which when complexed through mutual binding to a common ligand, are capable of actuating, directly or indirectly, the desired event.
- This invention encompasses recombinant DNA constructs encoding those chimeric proteins; DNA vectors containing one or more of those constructs; the fusion proteins encoded by the foregoing constructs; cells, especially animal cells, transformed with (i.e., containing and capable of expressing) one or more of the DNA constructs described herein; small molecules (bivalent or multivalent multimerizing agents) which bind to and are capable of inducing multimerization of the chimeric protein molecules; and, methods for preparing and using the foregoing.
- this invention provides methods and materials for making and using genetically engineered cells which are responsive to the presence of rapamycin or to the presence of an analog, mimic or derivative of rapamycin (a "rapalog").
- the invention relies upon the introduction into cells of recombinant DNAs encoding a set of fusion proteins which are capable of complexing with each other in the presence of rapamycin or a rapalog. Contacting such genetically engineered cells with rapamycin or a suitable rapalog results in complexation of the fusion proteins and the initiation of a biological response.
- One of the fusion proteins contains one or more copies of an FKBP:rapamycin binding (FRB) domain and at least one heterologous protein domain.
- FKBP FKBP:rapamycin binding
- the second fusion protein contains one or more copies of a domain derived from an FKBP protein which is capable of binding to rapamycin or a rapalog and forming a complex with an FRB- containing protein.
- the second fusion protein also contains at least one heterologous domain which mav be the same or different from a heterologous domain of the first fusion protein.
- FRB and FKBP domains for use in fusion proteins of this invention may be selected from naturally occurring proteins and may be variously modified, as is discussed in detail below. While FRB, FKBP and heterologous domains derived from various species may be used, human peptide sequences or variants thereof are preferred for human gene therapy applications.
- the FRB and FKBP domains serve as receptor (or "ligand-binding") domains and direct the complexation of the fusion proteins under the mediation of rapamycin or rapalog molecules.
- receptor or "ligand-binding” domains
- the nature of the biological response triggered by rapamycin- or rapalog-mediated complexation is determined by the heterologous domains of the fusion proteins.
- the heterologous domains are therefore also referred to as "action" domains.
- the two fusion proteins each contain at least one different heterologous domain, i.e., a heterologous domain not contained in the other fusion protein.
- one of the fusion proteins contains at least one DNA binding domain and the other fusion protein contains at least one transcription activation domain.
- Ligand-mediated association of the fusion proteins represents the formation of a transcription factor complex and leads to initiation of transcription of a target gene linked to a DNA sequence recognized by (i.e., capable of binding with) a DNA-binding domain on one of the fusion proteins.
- one of the fusion proteins contains at least one domain capable of directing the fusion protein to a particular cellular location such as the cell membrane, nucleus, etc.
- Localization domains which target the cell membrane include domains such as a myristoylation site or a transmembrane region of a receptor protein or other membrane-spanning protein.
- the other fusion protein contains a signalling domain capable, upon membrane localization and/or clustering, of activating a cellular signal transduction pathway.
- signalling domains include an intracellular domain of a growth factor or cytokine receptor, an apoptosis triggering domain such as the intracellular domain of FAS or TNF- R1, and domains derived from other intracellular signalling proteins such as SOS, Raf, lck, ZAP-70, etc.
- a number of signalling proteins are disclosed in PCT/US94/01617 (see e.g. pages 23 - 26).
- each of the fusion proteins contains at least one FRB domain and at least one FKBP domain, as well as one or more heterologous domains.
- Such fusion proteins are capable of homodimerization in the presence of rapamycin or a rapalog.
- domains containing peptide sequence endogenous to the host cell are preferred.
- domains of human origin are of particular interest.
- Recombinant DNA molecules encoding the fusion proteins are also provided, as are vectors capable of directing their expression, particularly in eukaryotic cells, of which yeast and animal cells are of particular interest.
- the recombinant DNA molecules which encode them are capable of selectively hybridizing (a) to a DNA molecule encoding a given fusion protein's ligand- binding domain (FRB domain or FKBP domain) or a protein containing such a domain and (b) to a DNA molecule encoding the heterologous domain or a protein from which the heterologous protein domain was derived.
- DNAs are also encompassed which would be capable of so hybridizing but for the degeneracy of the genetic code.
- DNA sequences encoding the chimeric proteins of this invention, and vectors capable of directing their expression in eukaryotic cells one may genetically engineer cells for a number of important uses. To do so, one first provides an expression vector or DNA construct for directing the expression in a eukaryotic (preferably animal) cell of the desired chimeric protein and then introduces the recombinant DNA into the cells in a manner permitting DNA uptake and expression of the introduced DNA in at least a portion of the cells.
- a eukaryotic preferably animal
- One object of this invention is thus to provide an animal cell containing recombinant DNAs encoding two fusion proteins as described herein.
- One of the fusion proteins is capable of binding to rapamycin or a rapalog and contains at least one FKBP domain and at least one domain heterologous thereto.
- the second fusion protein contains at least one FRB domain and at least one domain heterologous thereto and is capable of forming a tripartite complex with the first fusion protein and one or more molecules of rapamycin or a rapalog.
- one or more of the heterologous domains present on one of the fusion proteins are also present on the other fusion protein, i.e., the two fusion proteins have one or more common heterologous domains.
- each fusion protein contains one or more different heterologous domains.
- a specific object of this invention is to provide animal cells engineered such that contacting the cells with rapamycin or a rapalog leads to transcription of a target gene.
- Such cells contain, in addition to recombinant DNAs encoding the two fusion proteins, a target gene construct which comprises a target gene operably linked to a DNA sequence which is responsive to the presence of a complex of the fusion proteins with rapamycin or a rapalog.
- the cells are responsive to contact with a rapalog which binds to the FKBP fusion protein and FRB fusion protein with a detectable preference over binding to endogenous FKBP or FRB-containing proteins of the host cell.
- Another specific object of this invention is to provide animal cells engineered such that contacting the cells with rapamycin or a rapalog leads to the initiation of cell death.
- at least one of the heterologous domains on at least one of the fusion proteins is a domain such as the intracellular domain of FAS or TNF-R1, which, upon clustering, triggers apoptosis of the cell.
- Another specific object of this invention is to provide animal cells engineered such that contacting the cells with rapamycin or a rapalog stimulates cell growth,
- At least one of the heterologous domains on at least one of the fusion proteins is a domain such as the intracellular domain of a receptor for a hormone which mediates cell growth, differentiation or proliferation.
- Cell growth, differentiation and /or proliferation follow clustering of the receptor intracellular signalling domains. Such clustering occurs in nature following hormone binding, and in engineered cells of this invention following contact with rapamycin or a rapalog.
- Cells of human origin are preferred for human gene therapy applications, although cell types of various origins (human or other species) may be used, and may, if desired, be encapsulated within a biocompatible material for use in human subjects.
- Another object of the invention is to provide materials and methods for producing the foregoing engineered cells. This object is met by providing recombinant DNAs encoding the fusion proteins, together with any ancillary recombinant DNAs such as a target gene construct, and introducing the recombinant DNAs into the host cells under conditions permitting DNA uptake by cells. Such transfection may be effected ex vivo, using host cells maintained in culture. Cells that are engineered in culture may
- a host organism preferably a human or non-human mammal, which is responsive (as described herein) to the presence of rapamycin or a rapalog.
- transfection may be effected in vivo, using host cells present in a human or non-human host organism.
- the DNA molecules are introduced directly into the host organism under conditions permitting uptake of the DNA by one or more of the host organism's cells.
- This approach thus constitutes an alternative method for providing a host organism, preferably a human or non-human mammal, which is responsive (as described herein) to the presence of rapamycin or a rapalog.
- Various materials and methods for the introduction of DNA into cells in culture or in whole organisms are known in the art and may be adapted for use in practicing this invention.
- a method for multimerizing the fusion proteins of this invention by contacting cells engineered as described herein with an effective amount of rapamycin or a suitable rapalog permitting the rapamycin or rapalog to form a complex with the fusion proteins.
- multimerization of the fusion proteins triggers transcription of a target gene
- this constitutes a method for activating the expression of the target gene.
- the fusion proteins contain one or more signalling domains, this constitutes a method for activating a cellular signal transduction pathway.
- the fusion proteins contain one or more domains capable upon clustering of triggering apoptoric cell death
- these methods may be carried out in cell culture or in whole organisms, including human patients.
- the rapamycin or rapalog is added to the culture medium.
- the rapamycin or rapalog (which may be in the form of a pharmaceutical or veterinary composition) is administered to the whole organism, e.g., orally, parenterally, etc.
- the dose or rapamycin or rapalog administered to an animal is below the dosage level that would cause undue
- a further object of this invention is to provide kits for use in the genetic engineering of cells or human or non-human animals as described herein.
- One such kit contains recombinant DNA constructs encoding a pair of fusion proteins of this invention.
- the recombinant DNA constructs will generally be in the form of eukaryotic expression vectors suitable for introduction into animal cells and capable of directing the expression of the fusion proteins therein.
- the kit may also contain a sample of rapamycin or a rapalog capable of forming a complex with the encoded fusion proteins.
- the kit may further contain a multimerization antagonist such as FK506 or some other compound capable of binding to one of the fusion proteins but incapable of forming a complex with both.
- the recombinant DNA constructs encoding the fusion proteins will contain a cloning site in place of DNA encoding one or more of the heterologous domains, thus permitting the practitioner to introduce DNA encoding a heterologous domain of choice.
- the kit may also contain a target gene construct containing a target gene or cloning site linked to a DNA sequence responsive to the presence of the complexed fusion proteins, as described in more detail elsewhere.
- FIG. 1 (A) Rapamycin-dependent dimerization of a FKBP chimera containing a DNA- binding domain with a FRAP chimera containing a transcription activation domain is depicted. (B) Schematic diagrams of representative plasmids used in some of the experimental examples. Transcription factor fusion proteins were produced under the control of the human cytomegalovirus (hCMV) immediate early promoter and enhancer. "E” represents an epitope tag and "N” represents the SV40 T antigen nuclear
- the reporter genes consisted of a minimal hCMV promoter devoid of enhancer sequences flanked by 12 tandemly reiterated binding sites for ZFHD1.
- Figure 2 depicts various FRAP-derived and FKBP-derived domains of fusion proteins of the invention in schematic form, together with rapamycin and rapamycin analogs.
- Figure 3 compares results obtained in transcription assays using constructs with various multiples and configurations of FRAP and FKBP domains.
- Figure 4 depicts a dose-response curve for the rapamycin-dependent gene transcription as measured by production of secreted alkaline phosphatase activity at various rapamycin concentrations in transiently transfected cells (A, B) and in stably transfected cells (C, D).
- Plasmids encoding ZFHD1-3FKBP and lFRB-p65(361-450) were transfected into HT1080 cells with the pZHWTx12-CMV-SEAP reporter gene. SEAP activity secreted into the growth medium was measured following incubation with indicated concentration of rapamycin.
- B The indicated plasmids (-) were omitted and replaced with empty expression vector alone. SEAP activity was measured following incubation with or without 10 nM rapamycin as indicated. Transfections were performed in quadruplicate and mean values (in relative units) ⁇ standard deviation plotted.
- FIG. 1 Schematic diagram of an IRES-containing plasmid used to express both 1FRB-p65(361-550) and ZFHD1-3FKBP from a single transcript.
- Bottom pCGNN-1FRB-p65(361-550)-IRES-ZFHD1-3FKBP was transfected, along with a plasmid conferring resistance to zeocin, into HT1080 cells already containing the pLH- ZHWTx12-IL2-SEAP reporter gene stably integrated. Pools of zeocin-resistant clones, HT23 cells, were incubated with the indicated concentration of rapamycin and SEAP activity secreted into the growth medium measured.
- Figure 5 depicts the results of rapamycin-dependent gene transcription experiments with various configurations of transcription factor fusion proteins. The experiments were aimed at determining an optimal configuration for rapamycin-binding domains in this system.
- HT1080 cells were transfected with plasmids encoding the indicated ZFHD1- FKBP and FRB-p65(450-550) fusion proteins. Transfected cells were incubated with medium containing 10 nM rapamycin and SEAP activity measured. Transfections were performed in triplicate and mean values (in relative units) ⁇ standard deviation are plotted. Immunoblot analysis showed that all fusions were expressed at similar levels with the exception of ZFHD1-1FKBP which was a pressed at higher levels.
- Figure 7 demonstrates the ability of HT1080 cells, engineered as described in Example 4 (see also Figure 6) and transplanted into mice, to produce hGH for prolonged periods of time following intravenous rapamycin administration.
- Mice were treated with 10.0 mg/kg rapamycin following transplantation of transfected HT1080 cells.
- Figure 8 demonstrates the ability of transplanted engineered cells to be restimulated by a second rapamycin administration after the effects of the first administration have dissipated.
- the flat, horizontal line designated as "estimated trough rapamycin concentrations” represent the steady state trough concentrations of rapamycin that were calculated from the rapamycin pharmacokinetics and the 16 hour dosing interval used in the present study.
- the descending line which begins at 80 hours, represents the estimated circulating rapamycin concentrations which show an elimination half-life of 4.6 hours.
- Figure 9 shows the results of analysis of various rapamycin C24 oximes (prepared as described in Example 5) for FKBP binding affinity using the competitive FP assay described in Example 6. Results show the mean ⁇ SD of two independent experiments (the C24 methyl oxime plot shows a single experiment)
- Figure 12 depicts illustrative examples of configurations useful for rapamycin-dependent control of signal transduction processes.
- M/E denotes a myristoylation motif or extracellular receptor domain(s) that locate the chimeric constructs in the membrane;
- EFF denotes effector domains that elicit a cellular response upon oligomerization (for example, a polypeptide including the death domain of FAS). Both configurations can be used to homodimerize or heterodimerize effector domains (heterodimerization is illustrated in (a) by the designations EFF and EFF'). Note that many other possible configurations can be envisaged: in particular, the order and number of each domain can be varied as appropriate.
- Figure 13 shows reduction in survival- of cells expressing mixed chimeric proteins (a,b) comprising a myristoylation domain, an FRB domain from human FRAP, an FKBP domain from hFKBP12, and a portion of the intracellular domain of Fas, as described in Example 8.
- (c) illustrates control experiments using a chimera containing a myristoylation domain, two FKBP domains and a Fas intracellular domain, designed to dimerize in the presence of AP1428 but not in the presence of rapamcyin.
- Figure 14 shows a gel mobility shift assay of binding reactions containing approximately equal amounts of Lex-FKBP and Lex-FRAP proteins incubated with DNA containing a Lex binding site. In the absence of Rapamycin no specific binding is detectable ( Figure 14, Lane 1). The inclusion of increasing amounts of Rapamycin in the binding reaction results in the concomitant appearance of a specific complex in the mobility shift gel ( Figure 14, Lanes 2 - 4). These results support the conclusion that rapamycin promotes formation of a Lex-FKBP/Lex-FRAP heterodimer that is capable of binding to DNA containing a LexA target sequence. See Example 9 for additional details. Detailed Description of the Invention
- FRB domains are polypeptide regions (protein "domains"), typically of at least about 89 amino acid residues, which are capable of forming a tripartite complex with an FKBP protein and rapamvcin (or a rapalog). FRB domains are present in a number of naturally occurring proteins, including FRAP proteins (also referred to in the literature as "RAPT1” or RAFT”) from human and other species; yeast proteins including Tori and Tor2; and a Candida FRAP homolog.
- FRAP proteins also referred to in the literature as "RAPT1" or RAFT”
- yeast proteins including Tori and Tor2
- Candida FRAP homolog FRAP homolog
- FRB domains for use in this invention generally contain at least about 89-100 amino acid residues.
- Fig.2 of Chiu et al, supra displays a 160-amino acid span of human FRAP, murine FRAP, S. cerevisiae TOR1 and S. cerevisiae TOR2 encompassing the conserved FRB region.
- the FRB sequence selected for use in fusion proteins of this invention will span at least the 89-amino acid sequence Glu-39 through Lys/Arg- 127, as the sequence is numbered in that figure.
- the 89-amino acid sequence is numbered Glu-2025 through Lys-2113 in the case of human FRAP, Glu-1965 through Lys-2053 in the case of Tor2, and Glu-1962 through Arg-2050 in the case of Tori.
- An FRB peptide sequence for use in fusion proteins of this invention will be capable of binding to a complex of an FKBP protein bound to rapamycin or a rapalog (as may be determined by any means, direct or indirect, for detecting such binding), and may comprise a naturally occurring peptide sequence spanning the indicated 89-amino acid region of the proteins noted above or corresponding regions of homologous proteins; may contain up to about ten (preferably 1-5) amino acid substitutions, insertions or deletions within that region relative to the naturally occurring sequence; may be a peptide sequence encoded by a DNA sequence capable of selectively hybridizing to a DNA molecule encoding a naturally occurring FRB region; or may be encoded by a DNA sequence which would be capable, but for the degeneracy of the genetic code, of selectively hybridizing to a DNA molecule encoding a naturally occurring FRB region.
- FKBPs FK506 binding proteins
- FKBPs are the cytosolic receptors for macrolides such as FK506, FK520 and rapamycin and are highly conserved across species lines.
- FKBPs are proteins or protein domains which are capable of binding to rapamycin or to a rapalog of this invention and further forming a tripartite complex with an FRB-containing protein.
- Information concerning the nucleotide sequences, cloning, and other aspects of various FKBP species is already known in the art, permitting the synthesis or cloning of DNA encoding the desired FKBP peptide sequence, e.g., using well known methods and PCR primers based on published sequences. See e.g. Staendart et al, 1990, Nature 346, 671-674 (human FKBP12); Kay,
- FKBP domains for use in this invention varies, depending on which FKBP protein is employed.
- An FKBP peptide sequence for use in fusion proteins of this invention will be capable of binding to rapamycin or a rapalog and participating in a tripartite complex with a FRB-containing protein (as may be determined by any means, direct or indirect, for detecting such binding), and may comprise a naturally occurring peptide sequence derived from the human FKBP12 protein (exemplified below) or a peptide sequence derived from another human FKBP, from a murine or other mammalian FKBP, or from some other animal, yeast or fungal FKBP; may contain up to about ten (preferably 1-5) amino acid substitutions, insertions or deletions within that region relative to the naturally occurring sequence; may be a peptide sequence encoded by a DNA sequence capable of selectively hybridizing to a DNA molecule encoding a naturally occurring FKBP or may be encoded by a DNA sequence which would be capable, but for the degeneracy of the genetic code, of selectively hybridizing to a DNA molecule en
- Capable of selectively hybridizing means that two DNA molecules are susceptible to hybridization with one another, despite the presence of other DNA molecules, under hybridization conditions which can be chosen or readily determined empirically by the practitioner of ordinary skill in this art.
- Such treatments include conditions of high stringency such as washing extensively with buffers containing 0.2 to 6 x SSC, and/or containing 0.1% to 1% SDS, at temperatures ranging from room temperature to 65-75°C. See for example F.M. Ausubel et al., Eds, Short Protocols in Molecular Biology, Units 6.3 and 6.4 (John Wiley and Sons, New York, 3d Edition, 1995).
- Recombinant chimeric and “fusion”, as those terms are used herein, indicate that the various component domains or sequences are mutually heterologous in the sense that they do not occur together in the same arrangement in nature. More specifically, the component portions are not found in the same continuous polypeptide or nucleotide sequence or molecule in nature, at least not in the same order or orientation or with the same spacing present in the chimeric protein or recombinant DNA molecule of this invention.
- oligomerization refers to the association of two or more proteins, mediated, in the practice of this invention, by the binding of each such protein to a common ligand.
- a tripartite complex comprising a protein containing an FRB domain, a protein containing an FKBP domain and a molecule of rapamycin is an example of dimerization.
- fusion proteins contain multiple copies of FRAP and/or FRB domains. Complexes of such proteins may contain more than one molecule of rapamycin or the rapalog and more than one copy of one or more of the constituent proteins.
- Such multimeric complexes are still referred to herein as tripartite complexes to indicate the presence of the three types of constituent molecules, even if one or more are represented by multiple copies.
- Rapalogs are compounds other than rapamycin, preferably of molecular weight below 5kD, more preferably below 2.5 kD, which are capable of binding with an FKBP fusion protein and of forming a complex with an FKBP fusion protein and an FRB fusion protein. Rapalogs of particular interest include compounds, other than rapamycin itself, of the formula:
- U is -H, -OR 1 , -OC(O)R 1 , -OC(O)NHR 1 , -SR 1 , -NHR 1 , -NHC(O)R 1 , -NH-SO 2 - R 1 or -R 2 ;
- Y is -OR 5 , -OC(O)R 5 or -OC(O)NHR 5 ;
- R 2 is substituted aryl or allyl or alkylaryl
- R 3 is H, -R 7 , -C(O)R 7 , -C(O)NHR 7 or C-28 / C-30 cyclic carbonate; and R 1 , R 4 , EP, R 6 and R 7 are independently selected from H, alkyl, alkylaryl or aryl, including the individual resolved stereoisomers as well as mixtures thereof.
- Rapalogs of special interest form complexes with proteins comprising naturally occurring human FKBP and FRB domains with measurably lower affinity than with proteins containing corresponding FKBP and FRAP domains in which one or both of the receptor domains contain at least one amino acid replacement, deletion or addition, as described herein.
- the chimeric proteins contain at least one "receptor” or “ligand binding” domain comprising peptide sequence derived from a FRAP or FKBP protein and at least one "action” domain, heterologous with respect to the receptor domain, but capable, upon oligomerization of the chimeric protein molecules, of triggering a cellular or biological response.
- the action domains of the chimeric proteins may be selected from a broad variety of protein domains capable of effecting a desired biological result upon oligomerization or clustering of the chimeric proteins.
- one action domain may comprise a localization domain capable of directing the chimeric protein to a particular cellular location (membrane, nucleus, other organelle, etc.), or a signalling domain, e.g. derived from an intracellular domain of a growth factor receptor or cytokine receptor and capable, upon clustering or multimerization, of initiating an intracellular signal transduction pathway leading to cell growth or proliferation, to the transcription of a desired gene, to cell death or to some other desired result.
- clustering of chimeric proteins containing an action domain derived from the intracellular portion of the T cell receptor CD3 zeta domain triggers transcription of a gene under the transcriptional control of the IL-2 promoter or derivatives thereof.
- the action domain comprises a domain derived from proteins such as the FAS antigen or TNF-alpha receptor (TNFalpha-R1), which are capable, upon oligomerization, of triggering apoptosis of the cell.
- the action domains comprise a DNA-binding domain such as GAL4 and a transcription activation domain such as
- VP16 paired such that oligomerization of the chimeric proteins represents assembly of a transcription factor complex which triggers transcription of a gene linked to a DNA sequence recognized by (capable of specific binding interaction with) the DNA binding domain.
- DNA constructs which encode the expression of these chimeric proteins are provided for use in the genetic engineering of the host cells.
- To produce genetically engineered cells of this invention one introduces into host cells recombinant DNA molecules which comprise the foregoing DNA constructs and are capable of directing the expresssion of the desired chimeras. Any desired accessory constructs are also introduced. This may be
- the modified cells may then be selected, separated from other cells and cultured, again by conventional methods.
- Engineered cells of this invention contain and are capable of expressing at least one DNA construct encoding such a chimeric protein.
- the cells contain and are capable of expressing DNA constructs encoding a pair of chimeric proteins of this invention which are capable of multimerizing in the presence of an appropriate ligand.
- the cells further contain a target gene construct containing a target gene under the expression control of a DNA element responsive to the multimerized chimeric proteins.
- the engineered cells may be transiently transfected or stably transformed with one or more of the introduced DNA molecules.
- Useful multimerizing ligands include rapamycin and rapalogs which are capable of dimerizing FRB and FKBP domains of this invention.
- Such compounds are bivalent ligands, i.e., are capable of binding to, and thus multimerizing, two or more of the chimeric protein molecules containing FRAP and FRB domains, respectively.
- the first recombinant DNA molecule encodes a chimeric protein comprising (i) at least one FKBP domain capable of binding to rapamycin or a rapalog and (ii) at least one protein ("action") domain heterologous with respect to at least one of such FKBP domain.
- Such chimeric proteins which are referred to simply as “FKBP chimeras", are capable of bir ing to rapamycin or a rapalog to form a complex, analogous to the complex formed by the binding of naturally occurring FKBP proteins such as FKBP12 to rapamycin.
- the second recombinant DNA molecule encodes a chimeric protein containing (i) at least one FRB domain capable of binding to the complex formed by the first chimeric protein and rapamycin or a rapalog and (ii) a protein ("action") domain heterologous with respect to at least one of such FRB domain.
- FRAP chimeras Pairs of FKBP chimeras and FRAP chimeras are capable of forming a tripartite complex with rapamycin or a rapalog, as can be detected by a variety of means, including e.g.
- Embodiments involving multiple FKBP and/or FRB domains per chimera are capable of forming higher order multimers in the presence of rapamycin or the rapalog. Some embodiments involve a recombinant DNA molecule encoding a "mixed" chimeric protein containing one or more FKBP domains, one or more FRB domains and one or more action domains heterologous with respect to at least one of the receptor domains.
- Mixed chimeras are capable of forming protein homodimers or homomultimers via mutual binding of mixed chimeric protein molecules to rapamycin or to a rapalog.
- Chimeric proteins containing one or more ligand-binding (i.e., receptor) domains and one or more action domains, e.g. for activation of transcription of a target gene, triggering cell death or other signal transduction pathway, cellular localization, etc., are disclosed in PCT/US94/01617, PCT/US94/08008 and Spencer et al, supra.
- the design and use of such chimeric proteins for ligand-mediated gene-knock out and for ligand- mediated blockade of gene expression or inhibition of gene product function are disclosed in PCT/US95/10591.
- Novel DNA binding domains and DNA sequences to which they bind which are useful in embodiments involving regulated transcription of a target gene are disclosed, e.g., in Pomeranz et al, 1995, Science 267:93-96.
- Those references provide substantial information, guidance and examples relating to the design, construction and use of DNA constructs encoding analogous chimeras, target gene constructs, multivalent ligands, and other aspects which may also be useful to the practitioner of the subject invention. See also PCT /US95/ 06722 (Mitotix, Inc.). The full contents of the foregoing documents are incorporated herein by reference.
- this invention permits one to activate the transcription of a desired gene, actuate apoptosis, or trigger other biological events in engineered cells in a rapamycin- or rapalog-dependent manner analogous to the systems described in PCT/US94/01617 and PCT/US94/08008 and other references cited above.
- the engineered cells preferably animal cells, may be growing or maintained in culture or may be present within whole organisms, as in the case of human gene therapy, transgenic animals, and other such applications.
- the rapamycin or rapalog multimerizing agent is administered to the cell culture or to the organism containing the engineered cells, as the case may be, in an amount effective to multimerize chimeric proteins containing the corresponding ligand-binding domains (as may be observed indirectly by monitoring target gene transcription, apoptosis or other biological process so triggered).
- the rapamvcin or rapalog mav be administered in a composition containing the multimerizing agent and one or more acceptable verterinary or pharmaceutical diluents and/or excipients.
- a compound which binds to one of the chimeric proteins but does not form tripartite complexes with both chimeric proteins may be used as a multimerization antagonist. As such it may be administered to the engineered cells, or to organisms containing them (preferably in a composition as described above in the case of administration to whole animals), in an amount effective for blocking or reversing the effect of the multimerizing agent, i.e. for preventing, inhibiting or disrupting
- FK506, FK520 or any of the many synthetic FKBP ligands which do not form tripartite complexes with FKBP and FRAP may be used as an antagonist.
- One aspect of this invention provides materials and methods for ligand- dependent, direct activation of transcription of a desired gene.
- a set of two or more different chimeric proteins, and corresponding DNA constructs capable of directing their expression is provided.
- One such chimeric protein contains as its action domain(s) one or more transcriptional activation domains.
- the other chimeric protein contains as its action domain(s) one or more DNA-binding domains ( Figure 1). Rapamycin or a rapalog of this invention is capable of binding to both chimeras to form a dimeric or multimeric complex thus containing at least one DNA binding domain and at least one transcriptional activating domain.
- Formation of such complexes leads to activation of transcription of a target gene linked to, and under the transcriptional control of, a DNA sequence to which the DNA-binding domain is capable of binding, as can be observed by monitoring directly or indirectly the presence or concentration of the target gene product.
- the DNA binding domain, and a chimera containing it binds to its recognized DNA sequence with sufficient selectivity so that binding to the selected DNA sequence can be observed (directly or indirectly) despite the presence of other, often numerous other, DNA sequences.
- binding of the chimera comprising the DNA-binding domain to the selected DNA sequence is at least two, more preferably three and even more preferably more than four orders of magnitude greater than binding to any one alternative DNA sequence, as measured by in vitro binding studies or by measuring relative rates or levels of transcription of genes associated with the selected DNA sequence as compared with any alternative DNA sequences.
- Cells which have been genetically engineered to contain such a set of constructs, together with any desired accessory constructs, may be used in applications involving ligand-mediated, regulated actuation of the desired biological event, be it regulated transcription of a desired gene, regulated triggering of a signal transduction pathway such as the triggering of apoptosis, or another event.
- expression of a target gene for instance, can be used for regulated production of a desired protein (or other gene product) encoded by the target gene.
- a desired protein or other gene product
- Such cells may be grown in culture by conventional means. Addition of the ligand to the culture medium containing the cells leads to expression of the target gene by the cells and production of the protein encoded by that gene. Expression of the gene and production of the protein can be turned off by withholding further multimerization agent from the media, by removing residual multimerization agent from the media, or by adding to the medium a multimerization antagonist reagent.
- Engineered cells of this invention can also be produced and /or used in vivo, to modify whole organisms, preferably animals, especially humans, e.g. such that the cells produce a desired protein or other result within the animal containing them. Such uses include gene therapy applications.
- Embodiments involving regulatable actuation of apoptosis provide engineered cells susceptible to ligand-inducible cell death. Such engineered cells can be eliminated from a cell culture or host organism after they have served their intended purposed (e.g. production of a desired protein or other product), if they have or develop unwanted properties, or if they are no longer useful, safe or desired. Elimination is effected by adding the rapamycin or rapalog to the medium or administering it to the host organism. In such cases, the action domains of the chimeras are protein domains such as the intracellular domains of the FAS antigen or TNF-R1 which upon oligomerization trigger apoptosis.
- This invention thus provides materials and methods for achieving a biological effect in cells in response to the addition of a multimerizing ligand.
- the method involves providing cells engineered as described herein and exposing the cells to the ligand.
- this invention provides a method for activating transcription of a target gene in cells.
- the method involves providing cells containing (a) DNA constructs encoding a set of chimeric proteins of this invention capable upon ligand-mediated multimerization of initiating transcription of a target gene and (b) a target gene linked to an associated cognate DNA sequence responsive to the multimerization event (e.g. a DNA sequence recognized, i.e., capable of binding with, a DNA-binding domain of a foregoing chimeric protein.
- the method involves exposing the cells to a multimerization ligand capable of binding to the chimeric proteins in an amount effective to result in expression of the target gene.
- exposing the cells to the ligand may be effected by adding the ligand to the culture medium.
- exposing them to the ligand is effected by administering the ligand to the host organism.
- the ligand may be administered to the host organism by oral, bucal, sublingual, transdermal, subcutaneous, intramuscular, intravenous, intra-joint or inhalation administration in an appropriate vehicle therefor.
- the ligand-mediated biological event may be activation of a cellular function such as signal transduction leading to cell growth, cell proliferation, gene transcription, or apoptosis; deletion of a gene of interest, blockade of expression of a gene of interest, or inhibition of function of a gene product of interest; direct transcription of a gene of interest; etc.
- This invention further encompasses a pharmaceutical composition
- a pharmaceutical composition comprising rapamycin or a rapalog of this invention in admixture with a pharmaceutically acceptable carrier and optionally with one or more pharmaceutically acceptable excipients.
- Such pharmaceutical compositions can be used to promote multimerization of chimeras of this invention in engineered cells in whole animals, e.g. in human gene therapy applications to achieve any of the objectives disclosed herein.
- this invention provides a method for achieving any of those objectives, e.g. activation of transcription of a target gene (typically a heterologous gene for a therapeutic protein), cell growth or proliferation, cell death or some other selected biological event, in an animal, preferably a human patient, in need thereof and containing engineered cells of this invention.
- That method involves administering to the animal a pharmaceutical composition containing the rapamycin or rapalog by a route of administration and in an amount effective to cause multimerization of the chimeric proteins in at least a portion of the engineered cells. Multimerization may be detected indirectly by detecting the occurrence of target gene expression; cell growth, proliferation or death; or other objective for which the chimeras were designed and the cells genetically engineered.
- This invention further encompasses a pharmaceutical composition
- a pharmaceutical composition comprising a multimerization antagonist of this invention in admixture with a pharmaceutically acceptable carrier and optionally with one or more pharmaceutically acceptable excipients for inhibiting or otherwise reducing, in whole or part, the extent of
- multimerization of chimeric proteins in engineered cells of this invention in a subject and thus for de-activating the transcription of a target gene, for example, or turning off another biological result of this invention.
- the use of the multimerization reagents and of the multimerization antagonist reagents to prepare pharmaceutical compositions and achieve their pharmacologic results is encompassed by this invention.
- This invention also offers a method for providing a host organism, preferably an animal, typically a non-human mammal or a human subject, responsive to a host organism, preferably an animal, typically a non-human mammal or a human subject, responsive to a host organism, preferably an animal, typically a non-human mammal or a human subject, responsive to a host organism, preferably an animal, typically a non-human mammal or a human subject, responsive to a
- the method involves introducing into the organism cells which have been engineered in accordance with this invention, i.e.
- the engineered cells may be encapsulated using any of a variety of materials and methods before being introduced into the host organism.
- a host organism e.g. a mammal
- kits for producing cells responsive to rapamycin or a rapalog of this invention contains one or more DNA constructs encoding and capable of directing the expression of chimeras which, upon ligand-mediated
- the kit may contain a quantity of an oligomerizing ligand (rapamycin or a rapalog) capable of multimerizing the chimeric protein molecules encoded by the DNA construct(s) of the kit, and may contain in addition a quantity of a multimerization antagonist.
- the kit may further contain a DNA construct encoding a target gene (or cloning site) linked to a cognate DNA sequence which is recognized by the dimerized chimeric proteins permitting transcription of a gene linked to that cognate DNA sequence in the presence of multimerized chimeric protein molecules.
- the DNA constructs will preferably be associated with one or more selection markers for convenient selection of transfectants, as well as other conventional vector elements useful for replication in prokaryotes, for expression in eukaryotes, and the like.
- the selection markers may be the same or different for each different DNA construct, permitting the selection of cells which contain each such DNA construct(s).
- the accessory construct for introducing into cells a target gene in association with a cognate DNA sequence may contain a cloning site in place of a target gene.
- a kit constaining such a construct permits the engineering of cells for regulatable expression of a gene to be provided by the practitioner.
- kits of this invention may contain one or two (or more) DNA constructs for chimeric proteins in which one or more contain a cloning site in place of the
- kit may optionally include other elements as described above, e.g. DNA construct for a target gene with or without a cognate DNA sequence for a pre-selected DNA binding domain.
- kits may also contain positive control cells which were stably transformed with constructs of this invention such that they express a reporter gene (for CAT, beta-galactosidase or any conveniently detectable gene product) in response to exposure of the cells to the ligand.
- a reporter gene for CAT, beta-galactosidase or any conveniently detectable gene product
- Reagents for detecting and /or quantifying the expression of the reporter gene may also be provided.
- the FKBP chimeric protein comprises at least one ligand-binding domain containing all or part of the peptide sequence of an FKBP and at least one heterologous action domain.
- This chimeric protein must be capable of binding to rapamycin or a rapalog, preferably with a Kd value below about 100 nM, more preferably below about 10 nM and even more preferably below about 1 nM, as measured by direct binding measurement (e.g. fluorescence quenching), competition binding measurement (e.g. versus FK506), inhibition of FKBP enzyme activity (rotamase), or other assay methodology.
- the chimeric protein will contain one or more protein domains comprising peptide sequence corresponding to that of FKBP12, e.g.
- That peptide sequence may be modified to adjust the binding specificity, usually with replacement, insertion or deletion of 10 or fewer, preferably 5 or fewer, amino acid residues. Such modifications are elected in certain embodiments to yield one or both of the following binding profiles: (a) binding of a rapalog to the modified FKBP domain, or chimera containing it, preferably at least one, and more preferably at least two, and even more preferably three or four or more, orders of magnitude better (by any measure) than to FKBP12 or the FKBP endogenous to the host cells to be engineered; and (b) binding of the complex formed by the FKBP chimera with rapamycin or a rapalog to the second chimera (which, as discussed below, contains at least one FRB domain) preferably at least one, and more preferably at least two, and even more preferably at least three, orders of magnitude better (by any measure) than to the FRAP or other FRB-containing protein
- the FKBP chimera also contains at least one heterologous action domain, i.e., a protein domain containing non-FKBP peptide sequence.
- the action domain may be a DNA-binding domain, transcription activation domain, cellular localization domain, intracellular signal transduction domain, e.g. as described elsewhere herein or in
- the action domain is capable of directing the chimeric protein to a selected cellular location or of initiating a biological effect upon association or aggregation with another action domain, for instance, upon multimerization of proteins containing the same or different action domains.
- a recombinant DNA encoding such a protein will be capable of selectively hybridizing to a DNA encoding the parent FKBP protein, e.g. human FKBP12, or would be capable of such hybridization but for the degeneracy of the genetic code. Since these chimeric proteins contain an action domain derived from another protein, e.g. Gal4, VP16, FAS, CD3 zeta chain, etc., the recombinant DNA encoding the chimeric protein will also be capable of selectively hybridizing to a DNA encoding that other protein, or would be capable of such hybridization but for the degeneracy of the genetic code.
- FKBP chimeric proteins of this invention may contain one or more copies of one or more different ligand binding domains and one or more copies of one or more action domains.
- the ligand binding domain(s) may be N-terminal, C-terminal, or interspersed with respect to the action domain(s).
- Embodiments involving multiple copies of a receptor domain usually have 2 , 3 or 4 such copies.
- an FKBP chimera may contain 2, 3 or 4 FKBP domains.
- the various domains of the FKBP chimeras (and of the FRAP chimeras discussed below) are optionally separated by linking peptide regions which may be derived from one of the adjacent domains or may be heterologous.
- the FKBP fusion proteins comprise multiple copies of an FKBP domain containing amino acids 1-107 of human FKBP12, separated by the 2-amino acid linker Thr- Arg encoded by ACTAGA, the ligation product of DNAs digested respectively with the restriction endonucleases Spel and Xbal.
- the following table provides illustrative subsets of mutant FKBP domains based on the foregoing FKBP12 sequence:
- F36V designates a human FKBP12 sequence in which phenylalanine at position 36 is replaced by valine.
- F36V/F99A indicates a double mutation in which phenylalanine at positions 36 and 99 are replaced by valine and alanine, respectively.
- the second type of chimeric protein referred to as the "FRAP chimeric protein"
- comprises at least one FRB domain (which may comprise all or part of the peptide sequence of a FRAP protein or a variant thereof, as described elsewhere) and at least one heterologous protein ("action") domain.
- the FRB domain or a chimeric protein encompassing it, is encoded bv a DNA molecule capable of hybridizing selectively to a DNA molecule encoding a protein comprising a naturally occurring FRB domain, e.g. a DNA molecule encoding a human or other mammalian FRAP protein or one of yeast proteins, Tor-1 or Tor-2 or the previously mentioned Candida FRB-containing protein.
- FRB domains of this invention include those which are capable of binding to a complex of an FKBP protein and rapamycin or a rapalog. As disclosed in greater detail herein, rapalogs include compounds of the formula
- Y is -OR 5 , -OC(O)R 5 or
- Z O, -OR 6 , -NR 6 , -H, -NC(O)R 6 , or -OC(O)R 6 or -OC(O)NR 6 ;
- R 3 is H, -R 7 , -C(O)R 7 or -C(O)NHR 7 or C-28 / C-30 cyclic carbonate
- R 4 is H or alkyl
- R 1 , R 4 , R 5 , R 6 and R 7 are independently selected from H, alkyl, alkylaryl or aryl.
- the FRAP chimeric protein must be capable of binding to the complex formed by the FKBP chimera with rapamycin or a rapalog.
- the FRAP chimera binds to that complex with a Kd value below 200 ⁇ M, more preferably below 10 ⁇ M, as measured by conventional methods.
- the FRB domain will be of sufficient length and composition to maintain high affinity for a complex of rapamycin or a rapalog with the FKBP chimera.
- the FRB domain spans fewer than about 150 amino acids in length, and in some cases fewer than about 100 amino acids.
- One such region comprises a 133 amino acid region of human FRAP extending from Val 2012 through Tyr 2144 . See Chiu et al, 1994, Proc. Natl. Acad. Sci. USA 91:12574-12578.
- An FRB region of particular interest spans Glu 2025 through Gin 2114 of human FRAP and retains affinity for
- FKBP12-rapamycin In some embodiments Q2214 is removed from the 90-amino acid sequence rendering this an 89-amino acid FRB domain.
- the FRB peptide sequence may be modified to adjust the binding specificity, usually with replacement, insertion or deletion, of 10 or fewer, preferably 5 or fewer, amino acids. Such modifications are elected in certain embodiments to achieve a preference towards formation of the complex comprising one or more molecules of the FKBP chimera, FRAP chimera and rapamycin or a rapalog over formation of complexes of endogenous FKBP and FRAP proteins with the rapamycin/rapalog. Preferably that preference is at least one, and more preferably at least two, and even more preferably three, orders of magnitude (by any measure).
- a recombinant DNA encoding such a protein will be capable of selectively hybridizing to a DNA encoding a FRAP species, or would be capable of such
- chimeric proteins contain an action domain derived from another protein, e.g. Gal4, VP16, Fas, CD3 zeta chain, etc.
- the recombinant DNA encoding the chimeric protein will be capable of selectively hybridizing to a DNA encoding that other protein, or would be capable of such hybridization but for the degeneracy of the genetic code.
- FRB chimeras useful in the practice of this invention include those disclosed in the examples which follow, variants thereof in which one or more of the heterologous domains are replaced with alternative heterologous domains or supplemented with one or more additional heterologous domains, variants in which one or more of the FRB domains is a domain of non-human peptide sequence origin (such as Tor 2 or Candida for example), and variants in which the FRB domain is modified by amino acid substitution, replacement or insertion as described herein, so long as the chimera is capable of binding to a complex formed by an FKBP protein and rapamycin or a rapalog.
- variants thereof in which one or more of the heterologous domains are replaced with alternative heterologous domains or supplemented with one or more additional heterologous domains variants in which one or more of the FRB domains is a domain of non-human peptide sequence origin (such as Tor 2 or Candida for example), and variants in which the FRB domain is modified by amino acid substitution, replacement or insertion as
- An illustrative FRB fusion protein contains one or more 89-amino acid FRBs containing residues 2025-2113 of human FRAP, separated by the linker Thr-Arg formed by ligation of Spel-Xbal sites as mentioned previously. It should be appreciated that such restriction sites or linkers in any of the fusion proteins of this invention may be deleted, replaced or extended using conventional techniques such as site-directed mutagenesis.
- a third type of chimeric protein comprises one or more FKBP-derived ligand- binding (i.e., "receptor") domains and one or more heterologous action domains, but further contains one or more FRB domains as described for the FRAP chimeras.
- FKBP-derived ligand- binding i.e., "receptor”
- heterologous action domains but further contains one or more FRB domains as described for the FRAP chimeras.
- Mixed chimeric protein molecules are capable of forming homodimeric or homomultimeric protein complexes in the presence of rapamycin or a rapalog to which they bind.
- Embodiments involving mixed chimeras have the advantage of requiring the introduction into cells of a single recombinant DNA construct in place of two
- a recombinant DNA encoding a mixed chimeric protein will be capable of selectively hybridizing to a DNA encoding an FKBP protein, a DNA encoding FRAP, and a heterologous DNA sequence encoding the protein from which one or more action domains is derived (e.g. Gal4, VP16, Fas, CD3 zeta chain, etc.), or would be capable of such hybridization but for the degeneracy of the genetic code.
- a heterologous DNA sequence encoding the protein from which one or more action domains is derived e.g. Gal4, VP16, Fas, CD3 zeta chain, etc.
- heterologous action domains of the FKBP and FRAP chimeras are protein domains which, upon mutual association of the chimeric proteins bearing them, are capable of triggering (or inhibiting) DNA-binding and/or transcription of a target gene; actuating cell growth, differentiation, proliferation or apoptosis;
- Embodiments involving regulatable gene transcription involve the use of target gene constructs which comprise a target gene (which encodes a polypeptide, antisense RNA, ribozyme, etc. of interest) under the transcriptional control of a DNA element responsive to the association or multimerization the heterologous domains of the 1st and 2d chimeric proteins.
- the heterologous domains of the 1st and 2d chimeric proteins comprise a DNA binding domain such as Gal4 or a chimeric DNA binding domain such as ZFHD1, discussed below,and a transcriptional activating domain such as those derived from VP16 or p65, respectively.
- the multimerization of a chimeric protein containing such a transcriptional activating domain to a chimeric protein containing a DNA binding domain targets the transcriptional activator to the promoter element to which the DNA binding domain binds, and thus activates the transcription of a target gene linked to that promoter element.
- the transcription activation domain or substituting a repressor domain (see PCT/US94/01617) in place of a transcription activation domain provides an analogous chimera useful for inhibiting transcription of a target gene.
- Composite DNA binding domains and DNA sequences to which they bind are disclosed in Pomerantz et al, 1995, supra, the contents of which are incorporated herein by reference. Such composite DNA binding domains may be used as DNA binding domains in the practice of this invention, together with a target gene construct containing the cognate DNA sequences to which the composite DBD binds.
- the heterologous domains of the chimeras are effector domains of signaling proteins which upon aggregation or multimerization trigger the activation of transcription under the control of a responsive promoter.
- the signaling domain may be the intracellular domain of the zeta subunit of the T cell receptor, which upon aggregation, triggers transcription of a gene linked to the IL-2 promoter or a derivative thereof (e.g. iterated NF-AT binding sites).
- the heterologous domains are protein domains which upon mutual association are capable of triggering cell death.
- Examples of such domains are the intracellular domains of the Fas antigen or of the TNF Rl.
- Chimeric proteins containing a Fas domain can be designed and prepared by analogy to the disclosure of PCT/US94/01617. Engineered receptor domains
- the FKBP and FRB domains may contain peptide sequence selected from the peptide sequences of naturally occurring FKBP and FRB domains.
- Naturally occurring sequences include those of human FKBP12 and the FRB domain of human FRAP.
- the peptide sequences may be derived from such naturally occurring peptide sequences but contain generally up to 10, and preferably 1-5, mutations in one or both such peptide sequences. As disclosed in greater detail herein, and as illustrated in Figure 2, such mutations can confer number of important features.
- an FKBP domain may be modified such that it is capable of binding a rapalog preferentially, i.e. at least one, preferably two.
- An FRB domain may be modified such that it is capable of binding a (modified or unmodified) FKBP:rapalog complex preferentially, i.e. at least one, preferably two, and even more preferably three orders of magnitude more effectively, with respect to the unmodified FRB domain.
- FKBP and FRB domains may be modified such that they are capable of forming a tripartite complex with a rapalog, or with rapamycin, preferentially, i.e.
- Figures 2A-E are presented for the purposes of illustration only; other related combinations of variously modified rapalogs and receptor domains bearing compensatory mutations are also encompassed by this invention and may be adapted to various applications.
- mutants are expressed by standard methods and their binding affinity for the rapalogs measured, for example by inhibition of rotamase activity, or by competition for binding with a molecule such as FK506, if the mutant retains appropriate activity/affinity.
- FKBPs in which one or more of the residues, Tyr26, Phe36, Asp37, Tyr82 and Phe99, are substituted with amino acids that have smaller side chains (such as Gly, Ala, Val, Met and Ser).
- rapalogs with modifications at C20 i.e., rapalogs in which R 4 is other than -H
- rapalogs bearing modifications at C24 bind preferentially to FKBPs in which one or more of Phe46, Phe48 and Val55 are replaced by other amino acids, again especially those with smaller side chains.
- rapalogs with modifications at C28 and /or C30 bind preferentially to FKBPs in which Glu54 is replaced by another amino acid, especially one with a smaller side chain.
- Glu54 is replaced by another amino acid, especially one with a smaller side chain.
- single or multiple amino acid substitutions may be made. Again, specific examples are noted in the previous table.
- An alternative to iterative engineering and testing of single or multiple mutants is to co-randomize structurally-identified residues that are or would be in contact with or near one or more rapalog or rapamycin substituents.
- a collection of polypeptides containing FKBP domains randomized at the identified positions is prepared e.g. using conventional synthetic or genetic methods. Such a collection represents a set of FKBP domains containing replacement amino acids at one or more of such positions. The collection is screened and FKBP variants are selected which possess the desired rapalog binding properties.
- rapalog substituent (bump).
- the construction is also envisaged of unbiased libraries containing random substitutions that are not based on structural considerations, to identify subtle mutations or combinations thereof that confer preferential binding to bumped rapalogs.
- mutagenesis schemes include alanine-scanning mutagenesis (Cunningham and Wells (1989) Science 244, 1081-1085), PCR misincorporation mutagenesis (see eg. Cadwell and Joyce,1992, PCR Meth. Applic. 2, 28-33), and 'DNA shuffling' (Stemmer, 1994, Nature 370, 389-391 and Crameri et al, 1996, Nature Medicine 2, 100-103). These techniques produce libraries of random mutants, or sets of single mutants, that are then searched by screening or selection approaches.
- an effective strategy to identify the best mutants for preferential binding of a given bump is a combination of structure-based and unbiased approaches. See Clackson and Wells, 1994, Trends Biotechnology 12, 173-184 (review).
- libraries in which key contact residues are randomized by PCR with degenerate oligonucleotides, but with amplification performed using error-promoting conditions to introduce further mutations at random sites.
- a further example is the combination of component DNA fragments from structure-based and unbiased random libraries using DNA shuffling.
- Screening of libraries for desirable mutations may be performed by use of a yeast
- an FRB- VP16 fusion may be introduced into one vector, and a library of randomized FKBP sequences cloned into a separate GAL4 fusion vector.
- Yeast co-transformants are treated with bumped rapamycin (i.e., rapalog), and those harboring complementary FKBP mutants are identified by for example beta-galactosidase or luciferase production (a screen), or survival on plates lacking an essential nutrient (a selection), as appropriate for the vectors used.
- bumped rapamycin i.e., rapalog
- those harboring complementary FKBP mutants are identified by for example beta-galactosidase or luciferase production (a screen), or survival on plates lacking an essential nutrient (a selection), as appropriate for the vectors used.
- the requirement for bumped rapamycin to bridge the FKBP-FRAP interaction is a useful screen to eliminate false positives.
- a further strategy for isolating modified ligand-binding domains from libraries of FKBP (or FRB) mutants utilizes a genetic selection for functional dimer formation described by Hu et. al. (Hu, J.C., et al. 1990. Science. 250:1400-1403; for review see Hu, J.C. 1995. Structure. 3:431-433).
- This strategy utilizes the fact that the bacteriophage lambda repressor cI binds to DNA as a homodimer and that binding of such homodimers to operator DNA prevents transcription of phage genes involved in the lyric pathway of the phage life cycle.
- Repressor protein comprises an amino terminal DNA binding domain (amino acids 1-92), joined by a 40 amino acid flexible linker to a carboxy terminal dimerization domain.
- the isolated N-terminal domain binds to DNA with low affinity due to inefficient dimer formation. High affinity DNA binding can be restored with heterologous dimerization domains such as the GCN4 "leucine zipper".
- Hu et al have described a system in which phage immunity is used as a genetic selection to isolate GCN4 leucine zipper mutants capable of mediating lambda repressor dimer formation from a large population of sequences (Hu et. al., 1990).
- lambda repressor-FRAP libraries bearing mutant FRAP sequences are transformed into E. coli cells expressing wildtype lambda repressor- FKBP protein. Plasmids expressing FRAP mutants are isolated from those colonies that survive lysis on bacterial plates containing high titres of lambda phage and "bumped" rapamycin compounds.
- the above strategy is repeated with lambda repressor-FKBP libraries bearing mutant FKBP sequences transformed into E. coli cells expressing wildtype lambda repressor-FRAP protein.
- a further alternative is to clone the randomized FKBP sequences into a vector for phage display, allowing in vitro selection of the variants that bind best to the rapalog.
- Affinity selection in vitro may be performed in a number of ways. For example, rapalog is mixed with the library phage pool in solution in the presence of recombinant FRAP tagged with an affinity handle (for example a hexa-histidine tag, or GST), and the resultant complexes are captured on the appropriate affinity matrix to enrich for phage displaying FKBP harboring complementary mutations.
- an affinity handle for example a hexa-histidine tag, or GST
- mutant FRB domains which bind preferentially to rapalogs containing modifications (i.e., are 'bumped') relative to rapamycin in the FRAP-binding effector domain. For example, one may obtain
- Exemplary mutations include Y2038H, Y2038L, Y2038V, Y2038A, F2039H, F2039L, F2039A, F2039V, D2102A, T2098A, T2098N, andT2098S.
- Examplary mutations include E2032A and E2032S.
- Proteins comprising an FRB containing one or more amino acid replacements at the foregoing positions, libraries of proteins or peptides randomized at those positions (i.e., containing various substituted amino acids at those residues), libraries randomizing the entire protein domain, or combinations of these sets of mutants are made using the procedures described above to identify mutant FRAPs that bind preferentially to bumped rapalogs.
- the affinity of candidate mutant FRBs for the complex of an FKBP protein complexed with a rapalog may be assayed by a number of techniques; for example binding of in vitro translated FRB mutants to GST-FKBP in the presence of drug (Chen et al. 1995. Proc. Natl. Acad. Sci. USA 92, 4947-4951); or ability to participate in a rapalog-dependent transcriptionally active complex with an appropriate FKBP fusion protein in a yeast two-hybrid assay.
- FRB mutants with desired binding properties may be isolated from libraries displayed on phage using a variety of sorting strategies.
- a rapalog is mixed with the library phage pool in solution in the presence of recombinant FKBP tagged with an affinity handle (for example a hexa-histidine tag, or GST), and the resultant complexes are captured on the appropriate affinity matrix to enrich for phage displaying FRAP harboring complementary mutations.
- an affinity handle for example a hexa-histidine tag, or GST
- FRB fusion protein that may vary in the various embodiments of this invention is the exact sequence of the FRB domain used. In some applications it may be preferred to use portions of an FRB which are larger than the minimal (89 amino acid) FRB domain. These include extensions N-terminal to residue Glu2025 (preferably extending to at least Arg2018 or Ile2021), as well as C-terminal extensions beyond position 2113, e.g. to position 2113, 2141 or 2174 or beyond), which may in some cases improve the stability of the folded FRB domain and/or the efficiency of expression.
- FRB sequence termini may be used include those in which a long linker is desired for steric reasons on one or both sides of the FRB domain, for example to accommodate the distortions of the polypeptide chain required for FRB-mediated protein-protein association at the cell membrane or on DNA. Conversely, in other applications short linkers on one or both sides of the FRB domain may be preferred or required to present the heterologous action domain(s) appropriately for biological function. In human gene therapy applications the use of naturally occurring human FRAP sequence for such linkers will generally be preferred to the introduction of heterologous sequences, or reduce the risk of provoking an immune response in the host organism.
- Some rapalogs especially rapalogs with modifications or substituents (relative to rapamycin) at positions believed to lie near the boundary between the FKBP binding domain and the FRAP binding domain, such as those on C28, C30, C7 and C24, possess reduced ability, relative to rapamycin, to form complexes with both mammalian FKBP and FRB domains, in particular, with those domains containing naturally occurring human peptide sequence. That reduced ability may be manifested as a reduced binding affinity as determined by any of the direct or indirect assay means mentioned herein or as reduced immunosuppressive activity as determined in an appropriate assay such as a T cell proliferation assay.
- iterative procedures may be used to identify pairs of mutant FKBPs and mutant FRBs that are capable of complexing with the rapalog more effectively than the corresponding domains containing naturally occurring human peptide sequence. For example, one may first identify a complementary modified FKBP domain capable of binding to the rapalog, as discussed previously, and then using this mutant FKBP domain as an affinity matrix in complex with the rapalog, one may select a complementary modified FRB domain capable of associating with that complex. Several cycles of such mutagenesis and screening may be performed to optimize the protein pair.
- mutant FKBP domains containing mutations that can affect the protein-protein interaction are of particular interest.
- Similar selection and screening approaches to those delineated previously can be used (i) to identify amino acid substitutions, deletions or insertions to an FKBP domain which measurably diminish the domain's ability to form the tripartite complex with rapamycin or rapalog and the endogenous FRB; (ii) to identify amino acid substitutions, deletions or insertions to an FRB domain which measurably diminish the domain's ability to form the tripartite complex with rapamycin or rapalog and the endogenous FKBP; and (iii) to select and /or otherwise identify compensating mutation(s) in the partner protein.
- suitable mutant FKBPs with diminished effectiveness in tripartite complex formation, we include mammalian, preferably human FKBP in which one or both of His87 and Ile90 are replaced with amino acids such as Arg, Trp, Phe, Tyr or Lys which contain bulky side chain groups; FRB domains, preferably containing mammalian, and more preferably of human, peptide sequence may then be mutated as described above to generate complementary variants which are capable of forming a tripartite complex with the mutant FKBP and rapamycin or a given rapalog.
- Illustrative FRB mutations which may be useful with H87W or H87R hFKBP12s include human FRBs in which Y2038 is replaced by V, S, A or L; F2039 is replaced by A; and/or R2042 is replaced by L, A or S.
- Illustrative FRB mutations which may be useful with I90W or I90R hFKBP12s include human FRBs in which K2095 is replaced with L, S, A or T.
- immunogenicity of a polypeptide sequence is thought to require the binding of peptides by MHC proteins and the recognition of the presented peptides as foreign by endogenous T-cell receptors. It may be preferable, at least in human gene therapy applications, to tailor a given foreign peptide sequence, including junction peptide sequences, to minimize the probability of its being immunologically presented in humans. For example, peptide binding to human MHC class I molecules has strict requirements for certain residues at key 'anchor' positions in the bound peptide: eg.
- HLA- A2 requires leucine, methionine or isoleucine at position 2 and leucine or valine at the Cterminus (for review see Stern and Wiley (1994) Structure 2, 145-251). Thus in engineering proteins in the practice of this invention, this periodicity of these residues is preferably avoided, especially in human gene therapy applications.
- the foregoing applies to all protein engineering aspects of the invention, including without limitation the engineering of point mutations into receptor domains, and to the choice or design of boundaries between the various protein domains.
- the chimeric proteins may contain as a heterologous domain a cellular localization domain such as a membrane retention domain.
- a membrane retention domain can be isolated from any convenient membrane-bound protein, whether endogenous to the host cell or not.
- the membrane retention domain may be a transmembrane retention domain, i.e., an amino acid sequence which extends across the membrane as in the case of cell surface proteins, induing many receptors.
- the transmembrane peptide sequence may be extended to span part or all of an extracellular and/or intracellular domain as well.
- the membrane retention domain may be a lipid membrane retention domain such as a myristoylation or palmitoylation site which permits association with the lipids of the cell surface membrane.
- Lipid membrane retention domains will usually be added at the 5' end of the coding sequence for N-terminal binding to the membrane and, proximal to the 3' end for C-terminal binding.
- Peptide sequences involving post-translational processing to provide for lipid membrane binding are described by Carr, et al, PNAS USA (1988) 79, 6128; Aitken, et al, FEBS Lett.
- An amino acid sequence of interest includes the sequence M-G-S-S-K-S-K-P-K-D-P-S-Q-R.
- Various DNA sequences can be used to encode such sequences in the various chimeric proteins of this invention.
- localization domains include organelle-targeting domains and sequences such as -K-D-E- L and -H-D-E-L which target proteins bearing them to the endoplasmic reticulum, as well as nuclear localization sequences which are particularly useful for chimeric proteins designed for (direct) transcriptional regulation.
- organelle-targeting domains and sequences such as -K-D-E- L and -H-D-E-L which target proteins bearing them to the endoplasmic reticulum, as well as nuclear localization sequences which are particularly useful for chimeric proteins designed for (direct) transcriptional regulation.
- nuclear localization sequences which are particularly useful for chimeric proteins designed for (direct) transcriptional regulation.
- Various cellular localization sequences and signals are well known in the art.
- tissue specific regulatory elements in the constructs for expression of the chimeric proteins and the application of regulated transcription to the expression of Cre recombinase as the target gene leading to the elimination of a gene of interest flanked by loxP sequences.
- Those features may be adapted to the subject invention.
- the various domains of the chimeric proteins be derived from proteins of the same species as the host cell.
- the heterologous domains (as well as the FKBP and FRB domains) be of human origin, rather than of bacterial, yeast or other non-human source.
- epitope tags may also be incorporated into chimeric proteins of this invention to permit convenient detection.
- the chimeric proteins be expressed in a cell-specific or tissue-specific manner. Such specificity of expression may be achieved by operably linking one ore more of the DNA sequences encoding the chimeric protein(s) to a cell-type specific transcriptional regulatory sequence (e.g.
- promoter /enhancer Numerous cell-type specific transcriptional regulatory sequences are known. Others may be obtained from genes which are expressed in a cell-specific manner.
- constructs for expressing the chimeric proteins may contain regulatory sequences derived from known genes for specific expression in selected tissues.
- an appropriate target gene construct is also used in the engineered cells.
- Appropriate target gene constructs are those containing a target gene and a cognate tran.
- xriptional control element such as a promoter and /or enhancer which is responsive to the multimerization of the chimeric proteins.
- that responsiveness may be achieved by the presence in the target gene construct of one or more DNA sequences recognized by the DNA-binding domain of a chimeric protein of this invention (i.e., a DNA sequence to which the chimeric protein binds).
- responsiveness may be achieved by the presence in the target gene construct of a promoter and /or enhancer sequence which is activated by an intracellular signal generated by multimerization of the chimeric proteins.
- the target gene is linked to and under the expression control of the IL-2 promoter region.
- This invention also provides target DNA constructs containing (a) a cognate
- DNA sequence e.g. to which a DNA-binding chimeric protein of this invention is capable of binding (or which is susceptible to indirect activation as discussed above), and (b) flanking DNA sequence from the locus of a desired target gene endogenous to the host cells.
- These constructs permit homologous recombination of the cognate DNA sequence into a host cell in association with an endogenous target gene.
- the construct contains a desired gene and flanking DNA sequence from a target locus permitting the homologous recombination of the target gene into the desired locus.
- a target construct may also contain the cognate DNA sequence, or the cognate DNA sequence may be provided by the locus.
- the target gene in any of the foregoing embodiments may encode for example a surface membrane protein (such as a receptor protein), a secreted protein, a cytoplasmic protein, a nuclear protein, a recombinase such as Cre, a ribozyme or an antisense RNA.
- a surface membrane protein such as a receptor protein
- a secreted protein such as a cytoplasmic protein
- a nuclear protein such as Cre
- a recombinase such as Cre
- a ribozyme such as a ribozyme or an antisense RNA.
- This invention encompasses a variety of configurations for the chimeric proteins.
- the chimeric proteins share an important characteristic: cells containing constructs encoding the chimeras and a target gene construct express the target gene at least one, preferably at least two, and more preferably at least three or four or more orders of magnitude more in the presence of the multimerizing ligand than in its absence. Optimally, expression of the selected gene is not observed unless the cells are or have been exposed to a multimerizing ligand.
- the chimeric proteins are capable of initiating a detectable level of transcription of target genes within the engineered cells upon exposure of the cells to the rapamycin or rapalog ligand, i.e., following multimerization of the chimeras.
- transcription of target genes is activated in genetically engineered cells of this invention following exposure of the cells to a rapamycin or rapalog ligand capable of multimerizing the chimeric protein molecules.
- genetically engineered cells of this invention contain chimeric proteins as described above and are responsive to the presence and /or concentration of a rapamycin or rapalog ligand which is capable of multimerizing those chimeric protein molecules.
- That responsiveness is manifested by the activation of transcription of a target gene.
- transcriptional activity can be readily detected by any conventional assays for transcription of the target gene.
- the biological response to ligand-mediated multimerization of the chimeras is cell death or other biological events rather than direct activation of transcription of a target gene.
- the animal cells may be insect, worm or mammalian cells. While various mammalian cells may be used, including, by way of example, equine, bovine, ovine, canine, feline, murine, and non-human primate cells, human cells are of particular interest.
- various types of cells may be used, such as hematopoietic, neural, glial, mesenchymal, cutaneous, mucosal, stromal, muscle (including smooth muscle cells), spleen, reticuloendothelial, epithelial, endothelial, hepatic, kidney, gastrointestinal, pulmonary, fibroblast, and other cell types.
- hematopoietic cells which may include any of the nucleated cells which may be involved with the erythroid, lymphoid or myelomonocytic lineages, as well as myoblasts and fibroblasts.
- stem and progenitor cells such as hematopoietic, neural, stromal, muscle, hepatic, pulmonary, gastrointestinal and mesenchymal stem cells
- the cells may be autologous cells, syngeneic cells, allogeneic cells and even in some cases, xenogeneic cells with respect to an intended host organism.
- the cells may be modified by changing the major histocompatibility complex ("MHC") profile, by inactivating ⁇ 2-microglobulin to prevent the formation of functional Class I MHC molecules, inactivation of Class II molecules, providing for expression of one or more MHC molecules, enhancing or inactivating cytotoxic capabilities by enhancing or inhibiting the expression of genes associated with the cytotoxic activity, or the like.
- MHC major histocompatibility complex
- specific clones or oligoclonal cells may be of interest, where the cells have a particular specificity, such as T cells and B cells having a specific antigen specificity or homing target site specificity.
- Constructs encoding the chimeric proteins and target genes of this invention can be introduced into the cells as one or more DNA molecules or constructs, in many cases in association with one or more markers to allow for selection of host cells which contain the construct(s).
- the constructs can be prepared in conventional ways, where the coding sequences and regulatory regions may be isolated, as appropriate, ligated, cloned in an appropriate cloning host, analyzed by restriction or sequencing, or other convenient means. Particularly, using PCR, individual fragments including all or portions of a functional unit may be isolated, where one or more mutations may be introduced using "primer repair", ligation, in vitro mutagenesis, etc. as appropriate.
- the construct(s) once completed and demonstrated to have the appropriate sequences may then be introduced into a host cell by any convenient means.
- the constructs may be incorporated into vectors capable of episomal replication (e.g. BPV or EBV vectors) or into vectors designed for integration into the host cells' chromosomes.
- the constructs may be integrated and packaged into non-replicating, defective viral genomes like Adenovirus, Adeno-associated virus (AAV), or Herpes simplex virus (HSV) or others, including retroviral vectors, for infection or transduction into cells.
- the construct may be introduced by protoplast fusion, electroporation, biolistics, calcium phosphate transfection, lipofection, microinjection of DNA or the like.
- the host cells will in some cases be grown and expanded in culture before introduction of the construct(s), followed by the appropriate treatment for introduction of the construct(s) and integration of the construct(s).
- the cells will then be expanded and screened by virtue of a marker present in the constructs.
- markers which may be used successfully include hprt, neomycin resistance, thymidine kinase, hygromycin resistance, etc., and various cell- surface markers such as Tac, CD8, CD3, Thy1 and the NGF receptor.
- homologous recombination one may generally use either ⁇ or O-vectors. See, for example, Thomas and Capecchi, Cell (1987) 51, 503-512; Mansour, et al., Nature (1988) 336, 348-352; and Joyner, et al., Nature (1989) 338, 153-156.
- the constructs may be introduced as a single DNA molecule encoding all of the genes, or different DNA molecules having one or more genes.
- the constructs may be introduced simultaneously or consecutively, each with the same or different markers.
- Vectors containing useful elements such as bacterial or yeast origins of replication, selectable and/or amplifiable markers, promoter /enhancer elements for expression in procaryotes or eucaryotes, and mammalian expression control elements, etc. which may be used to prepare stocks of construct DNAs and for carrying out transfections are well known in the art, and many are commercially available. Introduction of Constructs into Animals
- Cells which have been modified ex vivo with the DNA constructs may be grown in culture under selective conditions and cells which are selected as having the desired construct(s) may then be expanded and further analyzed, using, for example, the polymerase chain reaction for determining the presence of the construct in the host cells and /or assays for the production of the desired gene product(s).
- modified host cells Once modified host cells have been identified, they may then be used as planned, e.g. grown in culture or introduced into a host organism.
- the cells may be introduced into a host organism, e.g. a mammal, in a wide variety of ways.
- Hematopoietic cells may be administered by injection into the vascular system, there being usually at least about 10 4 cells and generally not more than about 10-10 cells.
- the number of cells which are employed will depend upon a number of circumstances, the purpose for the introduction, the lifetime of the cells, the protocol to be used, for example, the number of
- the number of cells will be at least about 10 4 and not more than about 109 and may be applied as a dispersion, generally being injected at or near the site of interest.
- the cells will usually be in a physiologically-acceptable medium.
- Cells engineered in accordance with this invention may also be encapsulated, e.g. using conventional biocompatible materials and methods, prior to implantation into the host organism or patient for the production of a therapeutic protein. See e.g. Hguyen et al, Tissue Implant Systems and Methods for Sustaining viable High Cell Densities within a Host, US Patent No. 5,314,471 (Baxter International, Inc.); Uludag and Sefton, 1993, J Biomed. Mater. Res.
- the cells may then be introduced in encapsulated form into an animal host, preferably a mammal and more preferably a human subject in need thereof.
- the encapsulating material is semipermeable, permitting release into the host of secreted proteins produced by the encapsulated cells.
- the semipermeable encapsulation renders the encapsulated cells immunologically isolated from the host organism in which the encapsulated cells are introduced.
- the cells to be encapsulated may express one or more chimeric proteins containing component domains derived from proteins of the host species and/or from viral proteins or proteins from species other than the host species.
- the chimeras may contain elements derived from GAL4 and VP16.
- the cells may be derived from one or more individuals other than the recipient and may be derived from a species other than that of the recipient organism or patient.
- adenovirus adeno-associated virus
- retroviruses which allow for transfection and, in some cases, integration of the virus into the host. See, for example, Dubensky et al. (1984) Proc. Natl. Acad. Sci. USA 81, 7529-7533; Kaneda et al., (1989) Science 243,375-378; Hiebert et al. (1989) Proc. Natl. Acad. Sci.
- the vector may be administered by injection, e.g. intravascularly or intramuscularly, inhalation, or other parenteral mode.
- Non-viral delivery methods such as administration of the DNA via complexes with liposomes or by injection, catheter or biolistics may also be used.
- the manner of the modification will depend on the nature of the tissue, the efficiency of cellular modification required, the number of opportunities to modify the particular cells, the accessibility of the tissue to the DNA composition to be introduced, and the like.
- an attenuated or modified retrovirus carrying a target transcriptional initiation region if desired, one can activate the virus using one of the subject transcription factor constructs, so that the virus may be produced and transfect adjacent cells.
- the DNA introduction need not result in integration in every case. In some situations, transient maintenance of the DNA introduced may be sufficient. In this way, one could have a short term effect, where cells could be introduced into the host and then turned on after a predetermined time, for example, after the cells have been able to home to a particular site. Multimerizing Agents
- Ligands suitable for use in this invention include rapamycin and rapamycin analogs, derivatives and mimics (rapalogs) that are capable of binding to an FKBP chimera and a FRAP chimera to form a tripartitie complex.
- Rapalogs of this invention which contain one or more substituents ("bumps") that diminish, and preferably substantially preclude, their binding to FKBP and /or FRAP proteins endogenous to the engineered cells are preferred.
- Mutant FKBPs of this invention may be obtained and screened for binding to a given rapalog as described in PCT/US94/01617 and
- rapalogs possess optional substituents which diminish, and preferably substantially preclude, their binding as FKBP complexes to FRAP but which do bind, complexed to (mutant) FKBP chimeric proteins, to (mutant) FRAP chimeras ( Figure 2). Rapalogs containing such bumps permit more selective binding to mutant FKBP chimeras and /or mutant FRAP chimeras without interference by endogenous pools of FKBP12 or FRAP. This is desirable for a number of applications, especially uses in whole organisms.
- a further advantage of diminished binding to endogenous FRAP is that such multimerizing agents, alone or in conjunction with endogenous FKBP12 or chimeras containing FKBP domains, will possess correspondingly diminished
- chimeric proteins comprising mutant FKBP domains which due to surface residue mutation are substantially precluded from ligand-dependent binding to endogenous FRAP, but which can bind in a ligand-dependent manner to chimeric proteins containing one or more FRB domains with compensatory mutations (Figure 2-E).
- Monomeric monovalent ligands such as those disclosed in PCT/US94/01617, as well as derivatized compounds described herein, which are capable of binding to one of the chimeric proteins but not effecting dimerization or higher order multimerization thereof (in view of the monovalent nature of the ligand) are multimerization antagonists.
- Rapalogs of particular interest bind to human FKBP12 and /or inhibit its rotamase activity at least about an order of magnitude less potently than any of FK506, FK520 or rapamycin.
- Such assays are well known in the art. See e.g. Holt, et al, j. Amer. Chem. Soc.,1993, 115, 9925-9938.
- the diminution in inhibitory activity may be as great as about 2 orders of magnitude, and in some cases will exceed about three orders of magnitude.
- Useful rapalog substituents include among others, alkyl, aryl, -O-alkyl, -O- aryl, substituted or unsubstituted amine, amide, carbamide and ureas, where alkyl and aryl are as defined elsewhere herein. See e.g. PCT/US94/01617 and PCT/US94/08008.
- Multimerization ligands of this invention specifically include rapamycin and rapalogs of the formula
- Y is -OR 5 , -OC(O)R 5 or -OC(O)NHR 5
- Z O, -OR 6 , -NR 6 , -H,
- R 3 is H, -R 7 , -C(O)R 7 or -C(0)NHR 7 or C-28 / C-30 cyclic carbonate
- R 4 is H or alkyl, where R 1 , R 4 , R 5 , R 6 and R 7 are independently selected from H, alkyl, alkylaryl or aryl.
- Rapalogs of this invention may contain substituents in any of the possible stereoisomeric orientations.
- Alkvl as the term is used herein, is intended to include saturated and
- hydrocarbons generally containing 1 - 8 contiguous aliphatic carbon atoms (e.g. methyl, ethyl, n-propyl, iso-propyl, cyclopropyl, n-butyl, iso-butyl, sec-butyl, t-butyl, cyclobutyl, n-pentyl, iso-pentyl, sec-pentyl, cyclopentyl, and so on), which are optionally substituted with one or more functional groups selected from the group consisting of hydroxy, C 1 -C 8 alkoxy, acyloxy, carbamoyl, amino, N-acylamino, keto, halo (chloro, bromo, fluoro or iodo), trihalomethyl, cyano, carboxyl, alkyl, cycloalkyl, aryl and heteroaryl, which functional groups may themselves (with the exception of hydroxy, halo and cyano groups)
- Aryl as the term is used herein, is intended to include stable cyclic, heterocyclic, polycyclic, and polyheterocyclic unsaturated C 3 -C 14 moieties (exemplified by but not limited to phenyl, biphenyl, naphthyl, pyridyl, furyl, thiophenyl, imidazoyl, pyrimidinyl, and oxazoyl) which may be substituted with one to five functional groups selected from the group consisting of hydroxy, C 1 -C 8 alkoxy, C 1 -C 8 branched or straight-chain alkyl, acyloxy, carbamoyl, amino, N-acylamino, nitro, halo, trifluoromethyl, cyano, and carboxyl, which functional groups may themselves (with the exception of hydroxy, halo, triflouromethyl and cyano groups) bear one or more of the foregoing functional groups.
- Heteroalkyl and heteroaryl refer to alkyl and aryl moieties respectively, which contain one or more of oxygen, sulfur, or nitrogen in place of one or more carbon atoms.
- ligands of particular interest include a compound other than rapamycin itself, a reduced form of rapamycin bearing a hydroxyl group at position 14 or a rapamycin derivative in which U is a C2-C8 straightchain, branched or cyclic aliphatic or alkoxyl moiety; an aryl or heteroaryl substituted alkyl or alkoxyl moiety; or an aryl, aryloxy, heteroaryl or heteroaryloxy moiety, where the aryl and heteroaryl moeity may be substituted or unsubstituted.
- rapamycin or a rapalog of this invention may be used in pharmaceutical compositions and methods for promoting formation of tripartite complexes of chimeric proteins of this invention in a mammal containing genetically engineered cells of this invention.
- the preferred method of such treatment or prevention is by administering to a mammal an effective amount of the compound to promote measurable formation of such complexes in the engineered cells, or preferably, to promote measurable actuation of the desired biological event triggered by such complexation, e.g. transcription of a target gene, apoptosis of engineered cells, etc.
- Rapamycin and the various rapalogs can exist in free form or, where appropriate, in salt form.
- Pharmaceutically acceptable salts and their preparation are well-known to those of skill in the art.
- the pharmaceutically acceptable salts of such compounds include the conventional non-toxic salts or the quaternary ammonium salts of such compounds which are formed, for example, from inorganic or organic acids of bases.
- the compounds of the invention may form hydrates or solvates. It is known to those of skill in the art that charged compounds form hydrated species when lyophilized with water, or form solvated species when concentrated in a solution with an
- This invention also relates to pharmaceutical compositions comprising a therapeutically (or prophylactically) effective amount of the compound, and a
- Carriers include e.g. saline, buffered saline, dextrose, water, glycerol, ethanol, and combinations thereof, and are discussed in greater detail below.
- the composition if desired, can also contain minor amounts of wetting or emulsifying agents, or pH buffering agents.
- the composition can be a liquid solution, suspension, emulsion, tablet, pill, capsule, sustained release formulation, or powder.
- the composition can be formulated as a suppository, with traditional binders and carriers such as triglycerides.
- Oral formulation can include standard carriers such as pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharine, cellulose, magnesium carbonate, etc. Formulation may involve mixing, granulating and compressing or dissolving the ingredients as appropriate to the desired preparation.
- the pharmaceutical carrier employed may be, for example, either a solid or liquid.
- Illustrative solid carrier include lactose, terra alba, sucrose, talc, gelatin, agar, pectin, acacia, magnesium stearate, stearic acid and the like.
- a solid carrier can include one or more substances which may also act as flavoring agents, lubricants, solubilizers, suspending agents, fillers, glidants, compression aids, binders or tablet-disintegrating agents; it can also be an encapsulating material.
- the carrier is a finely divided solid which is in admixture with the finely divided active ingredient.
- the active ingredient is mixed with a carrier having the necessary compression properties in suitable proportions ,and compacted in the shape and size desired.
- the powders and tablets preferably contain up to 99% of the active ingredient.
- Suitable solid carriers include, for example, calcium phosphate, magnesium stearate, talc, sugars, lactose, dextrin, starch, gelatin, cellulose, methyl cellulose, sodium carboxymethyl cellulose, polyvinylpyrrolidine, low melting waxes and ion exchange resins.
- Illustrative liquid carriers include syrup, peanut oil, olive oil, water, etc. Liquid carriers are used in preparing solutions, suspensions, emulsions, syrups, elixirs and pressurized compositions.
- the active ingredient can be dissolved or suspended in a pharmaceutically acceptable liquid carrier such as water, an organic solvent, a mixture of both or pharmaceutically acceptable oils or fats.
- the liquid carrier can contain other suitable pharmaceutical additives such as solubilizers, emulsifiers, buffers, preservatives, sweeteners, flavoring agents, suspending agents, thickening agents, colors, viscosity regulators, stabilizers or osmo-regulators.
- liquid carriers for oral and parenteral administration include water (partially containing additives as above, e.g. cellulose derivatives, preferably sodium carboxymethyl cellulose solution), alcohols (including monohydric alcohols and polyhydric alcohols, e.g. glycols) and their
- the carrier can also be an oily ester such as ethyl oleate and isopropyl myristate.
- Sterile liquid carders are useful in sterile liquid form compositions for parenteral administration.
- the liquid carrier for pressurized compositions can be halogenated hydrocarbon or other pharmaceutically acceptable propellant.
- Liquid pharmaceutical compositions which are sterile solutions or suspensions can be utilized by, for example, intramuscular, intraperitoneal or subcutaneous injection. Sterile solutions can also be administered intravenously.
- the compound can also be administered orally either in liquid or solid composition form.
- the carrier or excipient may include time delay material well known to the art, such as glyceryl monostearate or glyceryl distearate along or with a wax, ethylcellulose, hydroxypropylmethylcellulose, methylmethacrylate and the like.
- time delay material such as glyceryl monostearate or glyceryl distearate along or with a wax, ethylcellulose, hydroxypropylmethylcellulose, methylmethacrylate and the like.
- Tween 80 in PHOSAL PG-50 phospholipid concentrate with 1,2-propylene glycol, A. Nattermann & Cie. GmbH
- PHOSAL PG-50 phospholipid concentrate with 1,2-propylene glycol, A. Nattermann & Cie. GmbH
- a wide variety of pharmaceutical forms can be employed. If a solid carrier is used, the preparation can be tableted, placed in a hard gelatin capsule in powder or pellet form or in the form of a troche or lozenge. The amount of solid carrier will vary widely but preferably will be from about 25 mg to about 1 g. If a liquid carrier is used, the preparation will be in the form of a syrup, emulsion, soft gelatin capsule, sterile injectable solution or suspension in an ampule or vial or nonaqueous liquid suspension.
- a pharmaceutically acceptable salt of rapamycin or a rapalog may be dissolved in an aqueous solution of an organic or inorganic acid, such as a 0.3M solution of succinic acid or citric acid.
- an organic or inorganic acid such as a 0.3M solution of succinic acid or citric acid.
- acidic derivatives can be dissolved in suitable basic solutions. If a soluble salt form is not available, the compound is dissolved in a suitable cosolvent or combinations thereof.
- suitable cosolvents include, but are not limited to, alcohol, propylene glycol, polyethylene glycol 300, polysorbate 80, glycerin, polyoxyethylated fatty acids, fatty alcohols or glycerin hydroxy fatty acids esters and the like in concentrations ranging from 0-60% of the total volume.
- rapamycin or rapalog Various delivery systems are known and can be used to administer the rapamycin or rapalog, or the various formulations thereof, including tablets, capsules, injectable solutions, encapsulation in liposomes, microparticles, microcapsules, etc.
- Methods of introduction include but are not limited to dermal, intradermal, intramuscular, intraperitoneal, intravenous, subcutaneous, inrranasal, pulmonary, epidural, ocular and (as is usually preferred) oral routes.
- the compound may be administered by any convenient or otherwise appropriate route, for example by infusion or bolus injection, by absorption through epithelial or mucocutaneous linings (e.g., oral mucosa, rectal and intestinal mucosa, etc.) and may be administered together with other biologically active agents. Administration can be systemic or local.
- preferred routes of administration are oral, nasal or via a bronchial aerosol or nebulizer.
- the composition is formulated in accordance with routine procedures as a pharmaceutical composition adapted for intravenous administration to human beings.
- compositions for intravenous administration are solutions in sterile isotonic aqueous buffer.
- the composition may also include a solubilizing agent and a local anesthetic to ease pain at the side of the injection.
- the ingredients are supplied either separately or mixed together in unit dosage form, for example, as a lyophilized powder or water free concentrate in a hermetically sealed container such as an ampoule or sachette indicating the quantity of active agent.
- the composition is to be administered by infusion, it can be dispensed with an infusion bottle containing sterile pharmaceutical grade water or saline.
- an ampoule of sterile water for injection or saline can be provided so that the ingredients may be mixed prior to administration.
- Administration to an individual of an effective amount of the compound can also be accomplished topically by administering the compound(s) directly to the affected area of the skin of the individual.
- the compound is administered or applied in a composition including a pharmacologically acceptable topical carrier, such as a gel, an ointment, a lotion, or a cream, which includes, without limitation, such carriers as water, glycerol, alcohol, propylene glycol, fatty alcohols, triglycerides, fatty acid esters, or mineral oils.
- Topical carriers include liquid petroleum, isopropyl palmitate, polyethylene glycol, ethanol (95%), polyoxyethylene monolaurate (5%) in water, or sodium lauryl sulfate (5%) in water.
- Other materials such as anti-oxidants, humectants, viscosity stabilizers, and similar agents may be added as necessary.
- Percutaneous penetration enhancers such as Azone may also be included.
- the compound may be disposed within devices placed upon, in, or under the skin.
- Such devices include patches, implants, and injections which release the compound into the skin, by either passive or active release mechanisms.
- the compound may be administered to patients in need of such treatment in a daily dose range of about 1 to about 2000 mg per patient.
- the dose administered it is preferred that the dose administered be below that associated with undue immunosuppressive effects.
- the amount of compound which will be effective in the treatment or prevention of a particular disorder or condition will depend in part on the nature of the disorder or condition, and can be determined by standard clinical techniques.
- in vitro or in vivo assays may optionally be employed to help identify optimal dosage ranges.
- Effective doses may be extrapolated from dose-response curves derived from in vitro or animal model test systems.
- the precise dosage level should be determined by the attending physician or other health care provider and will depend upon well known factors, including route of administration, and the age, body weight, sex and general health of the individual; the nature, severity and clinical stage of the disease; the use (or not) of concomitant therapies; and the nature and extent of genetic engineering of cells in the patient.
- the invention also provides a pharmaceutical pack or kit comprising one or more containers filled with one or more of the ingredients of the pharmaceutical compositions of the invention.
- a pharmaceutical pack or kit comprising one or more containers filled with one or more of the ingredients of the pharmaceutical compositions of the invention.
- Optionally associated with such container(s) can be a notice in the form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceutical or biological products, which notice reflects approval by the agency of manufacture, use or sale for human administration.
- This invention is applicable to any situation that calls for expression of an exogenously-introduced gene embedded within a large genome.
- the desired expression level could be preset very high or very low.
- the system may be further engineered to achieve regulated or titratable expression. See e.g. PCT/US93/01617. In most cases, the inadvertant activation of unrelated cellular genes is undesirable.
- the following are non-limiting examples of applications of the subject invention.
- This invention is particularly well suited for achieving regulated expression of a therapeutic target gene in the context of human gene therapy.
- One example uses a pair of chimeric proteins (ont containing a FRAP-derived receptor domain, the other containing an FKBP-derived receptor domain), a dimerizing agent capable of dimerizing the chimeras and a target gene construct to be expressed.
- One of the chimeric proteins comprises a composite DNA-binding domain as described in Pomerantz et al, supra, as the heterologous action domain.
- the second chimeric protein comprises a transcriptional activating domain as the heterologous action domain.
- the rapamycin or rapalog dimerizing reagent is capable of binding to both chimeras and thus of dimerizing or oligomerizing the chimeras.
- DNA molecules encoding and capable of directing the expression of these chimeric proteins are introduced into the cells to be engineered.
- Contacting the engineered cells or their progeny with the dimerizing reagent leads to assembly of the transcription factor complex and hence to expression of the target gene.
- the design and use of similar components is disclosed in PCT/US93/01617.
- a composite DNA-binding domain, and DNA sequence encoding it in place of the alternative DNA-binding domains; a FRAP-derived receptor domain on one of the chimeras in place of an FKBP-derived receptor domain; and a monomeric rapamycin- type dimerizing agent in place of a dimeric dimerizing agent such as disclosed in the referenced patent document.
- the level of target gene expression should be a function of the number or concentration of chimeric transcription factor complexes, which should in turn be a function of the concentration of the dimerizing ligand.
- Experimental data discussed below evidences such dose (of dimerizing ligand)-responsive gene expression.
- the dimerizing ligand may be administered to the patient as desired to activate transcription of the target gene. Depending upon the binding affinity of the ligand, the response desired, the manner of administration, the half-life, the number of cells present, various protocols may be employed.
- the ligand may be administered parenterally or orally. The number of administrations will depend upon the factors described above.
- the ligand may be taken orally as a pill, powder, or dispersion; bucally; sublingually; injected intravascularly, intraperitoneally, intramuscularly, subcutaneously; by inhalation, or the like.
- the ligand (and monomeric antagonist compound) may be formulated using conventional methods and materials well known in the art for the various routes of administration. The precise dose and particular method of
- administration will depend upon the above factors and be determined by the attending physician or human or animal healthcare provider. For the most part, the manner of administration will be determined empirically.
- a monomeric compound which can compete with the dimerizing ligand may be administered.
- an antagonist to the dimerizing agent can be administered in any convenient way, particularly intravascularly, if a rapid reversal is desired.
- cells may be eliminated through apoptosis via signalling through Fas or TNF receptor as described elsewhere. See
- the particular dosage of the ligand for any application may be determined in accordance with the procedures used for therapeutic dosage monitoring, where maintenance of a particular level of expression is desired over an extended period of times, for example, greater than about two weeks, or where there is repetitive therapy, with individual or repeated doses of ligand over short periods of time, with extended intervals, for example, two weeks or more.
- a dose of the ligand within a predetermined range would be given and monitored for response, so as to obtain a time-expression level relationship, as well as observing therapeutic response. Depending on the levels observed during the time period and the therapeutic response, one could provide a larger or smaller dose the next time, following the response. This process would be iteratively repeated until one obtained a dosage within the therapeutic range.
- the ligand is chronically administered, once the maintenance dosage of the ligand is determined, one could then do assays at extended intervals to be assured that the cellular system is providing the appropriate response and level of the expression product.
- the system is subject to many variables, such as the cellular response to the ligand, the efficiency of expression and, as appropriate, the level of secretion, the activity of the expression product, the particular need of the patient, which may vary with time and circumstances, the rate of loss of the cellular activity as a result of loss of cells or expression activity of individual cells, and the like.
- a similar problem is encountered in the construction and use of "packaging lines" for the production of recombinant viruses for commercial (e.g., gene therapy) and experimental use.
- These cell lines are engineered to produce viral proteins required for the assembly of infectious viral particles harboring defective recombinant genomes.
- Viral vectors that are dependent on such packaging lines include retrovirus, adenovirus, and adeno-associated virus.
- the titer of the virus stock obtained from a packaging line is directly related to the level of production of the viral rep and core proteins. But these proteins are highly toxic to the host cells. Therefore, it has proven difficult to generate high-titer recombinant AAV viruses.
- This invention provides a solution to this problem, by allowing the construction of packaging lines in which the rep and core genes are placed under the control of regulatable transcription factors of the design described here.
- the packaging cell line can be grown to high density, infected with helper virus, and transfected with the recombinant viral genome. Then, expression of the viral proteins encoded by the packaging cells is induced by the addition of dimerizing agent to allow the production of virus at high titer.
- Biological research This invention is applicable to a wide range of biological experiments in which precise control over a target gene is desired. These include: (1) expression of a protein or RNA of interest for biochemical purification; (2) regulated expression of a protein or RNA of interest in tissue culture cells (or in vivo, via engineered cells) for the purposes of evaluating its biological function; (3) regulated expression of a protein or RNA of interest in transgenic animals for the purposes of evaluating its biological function; (4) regulating the expression of a gene encoding another regulatory protein, ribozyme or antisense molecule that acts on an endogenous gene for the purposes of evaluating the biological function of that gene.
- Transgenic animal models and other applications in which the components of this invention may be adapted include those disclosed in PCT/US95/10591.
- kits useful for the foregoing applications contain DNA constructs encoding and capable of directing the expression of chimeric proteins of this invention (and may contain additional domains as discussed above) and, in embodiments involving regulated gene transcription, a target gene construct containing a target gene linked to one or more transcriptioal control elements which are activated by the multimerization of the chimeric proteins.
- the target gene construct may contain a cloning site for insertion of a desired target gene by the practitioner.
- kits may also contain a sample of a dimerizing agent capable of dimerizing the two recombinant proteins and activating transcription of the target gene. Examples
- Example 1 Constructs encoding chimeric transcription factors A. Unless otherwise stated, all DNA manipulations described in this and other examples were performed using standard procedures (See e.g., F.M. Ausubel et al, Eds., Current Protocols in Molecular Biology (John Wiley & Sons, New York, 1994).
- GAL4 DNA binding domain the HSV VP16 activation domain, human T cell CD3 zeta chain intracellular domain or the intracellular domain of human FAS are disclosed in PCT/US94/01617.
- Additional DNA vectors for directing the expression of fusion proteins relevant to this invention were derived from the mammalian expression vector pCGNN (Attar, R.M. and Gilman, M.Z. 1992. MCB 12: 2432-2443). Inserts cloned as Xbal-BamHI fragments into pCGNN are transcribed under the control of the human CMV promoter and enhancer sequences (nucleotides -522 to +72 relative to the cap site), and are expressed with an optional epitope tag (a 16 amino acid portion of the H. influenzae hemaglutinin gene that is recognized by the monoclonal antibody 12CA5) and, in the case of transcription factor domains, with an N-terminal nuclear localization sequence (NLS; from SV40 T antigen).
- pCGNN a mammalian expression vector pCGNN
- Inserts cloned as Xbal-BamHI fragments into pCGNN are transcribed under the control of the human CMV promoter and enhance
- human thymus total RNA (Clontech)
- the appropriately-sized band was purified, digested with Xbal and Spel, and ligated into Xbal-Spel digested pCGNN-GAL4. This construct was confirmed by restriction analysis (to verify the correct orientation) and DNA sequencing and designated pCGNN-GAL4- 1FRB.
- Vectors were also constructed that encode larger fragments of FRAP, encompassing the minimal FRB domain (amino acids 2025-2113) but extending beyond it.
- PCR primers were designed that amplify various regions of FRAP flanked by 5' Xbal and 3' Spel sites as indicated below.
- fragment FRAPj was amplified by RT-PCR as described above, digested with Xbal and Spel, and ligated into Xbal-Spel digested pCGNN-GAL4.
- This construct, pCGNN-GAL4-FRAP i was analyzed by PCR to confirm insert orientation and verified by DNA sequencing. It was then used as a PCR substrate to amplify the other fragments using the primers listed.
- the new fragments were cloned as GAL4 fusions as described above to yield the constructs pCGNN-GAL4-FRAP a , pCGNN-GAL4-FRAP b etc, which were confirmed by DNA sequencing.
- Vectors encoding concatenates of two of the larger FRAP fragments, FRAPd and FRAPe, were generated by analogous methods to those used earlier.
- Xbal-BamHI fragments encoding FRAP d and FRAP e were isolated from pCGNN-GAL4-FRAP d and pCGNN-GAL4-FRAP e and ligated back into the same vectors digested with Spel and BamHI to generate pCGNN-GAL4-2FRAP d and pCGNN-GAL4-2FRAP e .
- the Xbal-BamHI fragments encoding 1, 2, 3 and 4 copies of FRB were recovered from the GAL4 fusion vectors and ligated into Xbal- BamHI digested pCGNN to yield pCGNN-lFRB, pCGNN-2FRB etc. These vectors were then digested with Spel and BamHI.
- An Xbal-BamHI fragment encoding amino acids 414-490 of VP16 was isolated from plasmid pCG-Gal4-VP16 (Das, G., Hinkley, C.S. and Herr, W.
- Zif268 sequences were amplified from a cDNA clone by PCR using primers 5'Xba/Zif and 3'Zif+G.
- Octl homeodomain sequences were amplified from a cDNA clone by PCR using primers 5'Not Oct HD and Spe/Bam 3'Oct.
- the Zif268 PCR fragment was cut with Xbal and Notl.
- the Octl PCR fragment was cut with Notl and BamHI.
- pCGNNZFHDl pCGNNZFHDl in which the cDNA insert is under the transcriptional control of human CMV promoter and enhancer sequences and is linked to the nuclear localization sequence from SV40 T antigen.
- the plasmid pCGNN also contains a gene for ampicillin resistance which can serve as a selectable marker.
- pCGNNZFHD1-FKBPx1 and pCGNNZFHDl-FKBPx3 were prepared containing one or three tandem repeats of human FKBP12 ligated as an Xbal-BamHI fragment between the Spel and BamHI sites of pCGNNZFHD1.
- a sample of pCGNNZFHDl-FKBPx3 has been deposited with the American Type Culture Collection under ATCC Accession No. 97399.
- the Xbal-BamHI fragments encoding 1, 2, 3 and 4 copies of FRB were recovered from the GAL4 fusion vectors and ligated into Spe-BamHI digested pCGNN- ZFHD1 to yield pCGNN-ZFHD1-1FRB, pCGNN-ZFHD1-2FRB etc. Constructs were verified by restriction analysis and /or DNA sequencing.
- FRAP fragments encoding extra sequence N- terminal to FRB were cloned as ZFHD1 fusions.
- Xbal-BamHI fragments encoding FRAP a , FRAP b , FRAP c , FRAP d and FRAP e were excised from the vectors pCGNN-GAL4- FRAP a , pCGNN-GAL4-FRAP b etc and ligated into Spel-BamHI digested pCGNNZFHD1 to yield the vectors pCGNN-ZFHDl-FRAP a , pCGNN-ZFHDl-FRAP b , etc.
- Vectors encoding fusions of ZFHD1 to 2, 3 and 4 C-terminal copies of FRAP e were also constructed by isolating Xbal-BamHI fragments encoding 2FRAP e , 3FRAP e and 4FRAP e from pCGNN-GAL4-2FRAP e , pCGNN-GAL4-3FRAP e and pCGNN-GAL4-4FRAP e and ligating them into Spel-BamHI digested pCGNN-ZFHD1 to yield the vectors pCGNN- ZFHD1-2FRAP e , pCGNN-ZFHD1-3FRAP e and pCGNN-ZFHD1-4FRAP e . All constructs were verified by restriction analysis.
- Vectors were also constructed that encode N-terminal fusions of FRB domain(s) with ZFHD1.
- Xbal-BamHI fragments encoding 1, 2, 3 and 4 copies of FRAP e were isolated from pCGNN-GAL4-1FRAP e , pCGNN-GAL4-2FRAP e etc and ligated into Xbal-BamHI digested pCGNN to yield the plasmids pCGNN-lFRAP e , pCGNN-2FRAP e etc.
- p65 human NF-kB p65 subunit
- Primers 9 p65/ 5' Xba
- 11 p65 3' Spe/Bam
- primers 10 p65/361 Xba
- 11 amplify the coding sequence for amino acids 361-550, both flanked by 5' Xbal and 3' Spel/BamHI sites.
- PCR products were digested with Xbal and BamHI and cloned into Xbal-BamHI digested pCGNN to yield pCGNN-p65(450-550) and pCGNN-p65(361- 550).
- the constructs were verified by restriction analysis and DNA sequencing.
- the 100 amino acid P65 transcription activation sequence is encoded by the following linear sequence:
- plasmids pCGNN-1FRB, pCGNN-2FRB etc were digested with Spel and BamHI.
- An Xbal-BamHI fragment encoding p65 (450-550) was isolated from pCGNN-p65(450-550) and ligated into the Spel-BamHI digested vectors to yield the plasmids pCGNN-1FRB-p65(450-550), pCGNN-2FRB-p65(450-550) etc.
- the construct pCGNN-lFRB-p65(361-550) was made similarly using an Xbal-BamHI fragment isolated from pCGNN-p65(361-550). These constructs were verified by restriction analysis.
- FRAP fragments encoding extra sequence C-terminal to FRB were cloned as p65 fusions.
- Xbal-BamHI fragments encoding FRAP a , FRAP b , FRAP f , FRAP g and FRAP h were excised from the vectors pCGNN- GAL4-FRAP a , pCGNN-GAL4-FRAP b etc and ligated into Xbal-BamHI digested pCGNN to yield the vectors pCGNN-FRAP a , pCGNN-FRAP b , etc.
- plasmids were then digested with Spel and BamHI, and a Xbal-BamHI fragment encoding p65 (amino acids 450-550) ligated in to yield the five vectors pCGNN-FRAP a -p65, pCGNN-FRAP b - p65, etc, which were verified by restriction analysis.
- Vectors encoding fusions of p65 to 1 and 3 N-terminal copies of FRAP e were also prepared by digesting pCGNN-lFRAP e and pCGNN-3FRAP e with Spel and BamHI. Xbal-BamHI fragments encoding p65(450-550) and p65(361-550) (isolated from pCGNN-p65(450-550) and pCGNN-p65(361-550)) were then ligated in to yield the vectors pCGNN-lFRAP e -p65(450-550), pCGNN-3FRAP e - ⁇ 65(450-550), pCGNN- lFRAP e -p65(361-550) and pCGNN-3FRAP e -p65(361-550).
- Plasmids pCGNN-p65(450-550) and pCGNN-p65(361-550) were digested with Spel and BamHI, and Xbal-BamHI fragments encoding 1 and 3 copies of FRAP e (isolated from pCGNN-GAL4-lFRAP e and pCGNN- GAL4-3FRAP e ) and 1 copy of FRB (isolated from P CGNN-GAL4-1FRB) ligated in to yield the plasmids pCGNN- P 65(450-550)-lFRAP e , pCGNN-p65(450-550)-3FRAP e , pCGNN-p65(361-550)-lFRAP e , pCGNN-p65(361-550)-3FRAP e
- primers 12 and 13 are used to amplify the entire coding region of FRAP.
- Primers 1 and 13, 6 and 13, and 5 and 13 are used to amplify three fragments encompassing the FRB domain and extending through to the C-terminal end of the protein (including the lipid kinase homology domain). These fragments differ by encoding different portions of the protein N-terminal to the FRB domain.
- RT-PCR is used as described above to amplify the regions from human thymus RNA, the PCR products are purified, digested with Xbal and Spel, ligated into Xbal-Spel digested pCGNN, and verified by restriction analysis and DNA sequencing.
- Non-coding nucleotides are indicated in lower case (S/X) and (X/S) indicate the result of a ligation event between the compatible products of digestion with Xbal and Spel, to produce a sequence that is cleavable by neither enzyme
- the internal ribosome entry sequence (IRES) from the encephalomyocarditis virus was amplified by PCR from pWZL-Bleo.
- the resulting fragment which was cloned into pBS-SK+ (Stratagene), contains an Xbal site and a stop codon upstream of the IRES sequence and downstream of it, an Ncol site encompassing the ATG followed by Spel and BamHI sites.
- the sequence around the initiating ATG of pCGNN-ZFHDl-3FKBP was mutated to an Ncol site and the Xbal site was mutated to a Nhel site using the oligonucleotides
- Retroviral vectors used to express transcription factor fusion proteins from stably integrated, low copy genes were derived from pSR ⁇ MSVtkNeo (Muller et al., MCB 11:1785-92, 1991) and pSR ⁇ MSV(Xba ⁇ ) (Sawyers et al., J. Exp. Med. 181:307-313, 1995).
- Unique BamHI sites in both vectors were removed by digesting with BamHI, filling in with Klenow and religating to produce pSMTN2 and pSMTX2, respectively.
- pSMTN2 expresses the Neo gene from an internal thymidine kinase promoter.
- a Zeocin gene (Invitrogen) will be cloned as a Nhel fragment into a unique Xbal site downstream of an internal thymidine kinase promoter in pSMTX2 to yield pSNTZ.
- This Zeocin fragment was generated by mutagenizing pZeo/SV (Invitrogen) using the following primers to introduce Nhel sites flanking the zeocin coding sequence.
- pSMTN2 contains unique EcoRI and Hindlll sites downstream of the LTR. To facilitate cloning of transcription factor fusion proteins synthesized as Xbal-BamHI fragments the following sequence was inserted between the EcoRI and Hindlll sites to create pSMTN3:
- the equivalent fragment is inserted into a unique EcoRI site of pSMTZ to create pSMTZ3 with the only difference being that the 3' Hindlll site is replaced by an EcoRI site.
- pSMTN3 and pSMTZ3 permit chimeric transcription factors to be cloned downstream of the 5' viral LTR as Xbal-BamHI fragments and allow selection for stable integrants by virtue of their ability to confer resistance to the antibiotics G418 or Zeocin respectively.
- pCGNN-ZFHDl-3FKBP was first mutated to add an EcoRI site upstream of the first amino acid of the fusion protein.
- An EcoRI-BamHI(blunted) fragment was then cloned into EcoRI- Hind ⁇ II(blunted) pSR ⁇ MSVtkNeo (ref. 51) so that ZFHD1-3FKBP was expressed from the retroviral LTR.
- effector plasmids containing Gal4 DNA binding domain fused to one or more copies of an FRB domain were co-transfected with a plasmid encoding three FKBP domains and a p65 activation domain (3xFKBP-p65) by transient transfection.
- the data shown in Fig 3A indicate that in this system, four copies of the FRB domain fused to the Gal4 DNA binding domain activated the stably integrated reporter gene more strongly than other corresponding fusion proteins with fewer FRB domains.
- HT1080 B cells wore grown in MEM supplemented with 10% Bovine Calf Serum. Approximately 4x105 cells /well in a 6 well plate ⁇ vere transiently transfected by Lipofection procedure as recommended by GIBCO, BRL. The DNA: Lipofectamine ratio used correspond to 1:6. Cells in each well recieved 1.9 ug of PUC 118 plasmid as carrier , 100 ng of pCGNNGal4F3 and 500 ng one of the following plasmids :pCGNNl, 2, 3 or 4 FRB-p65. Following transfection, 2 ml fresh media was added and supplemented with Rapamycin to the indicated concentration. After 24 hrs, 100 ul of the media was assayed for SEAP activity as described (Spencer et al, 1993).
- Fig. 5 Several different configurations for transcription factor fusion proteins were explored (Fig. 5).
- FKBP domains were fused to ZFHD1 and FRBs to p65, optimal levels of rapamycin-induced activation ocurred when there were multiple FKBPs fused to ZFHD1 and fewer FRBs fused to p65.
- the preference for multiple drug-binding domains on the DNA-binding protein may reflect the capacity of these proteins to recruit multiple activation domains and therefore to elicit higher levels of promoter activity.
- the presence of only 1 drug-binding domain on the activation domain should allow each FKBP on ZFHD to recruit one p65. Any increase in the number of FRBs on p65 would increase the chance that fewer activation domains would be recruited to ZFHD, each one linked my multiple FRB-FKBP interactions.
- HT1080 cells (ATCC CCL-121), derived from a human fibrosarcoma, were grown in MEM supplemented with non-essential amino acids and 10% Fetal Bovine Serum. Cells plated in 24-well dishes (Falcon, 6 x 10 4 cells/well) were transfected using
- Lipofectamine under conditions recommended by the manufacturer (GIBCO/BRL). A total of 300 ng of the following DNA was transfected into each well: 100 ng ZFHDx12- CMV-SEAP reporter gene, 2.5ng pCGNN-ZFHDl-3FKBP or other DNA binding domain fusion, 5 ng pCGNN-1FRB-p65(361-550) or other activation domain fusion and 192.5 ng pUC118. In cases where the DNA binding domain or activation domain were omitted an equivalent amount of empty pCGNN expression vector was substituted. Following lipofection (for 5 hours) 500 ⁇ l medium containing the indicated amounts of rapamycin was added to each well.
- transiently transfected HT1080 cells for injection into mice (See below), cells in 100 mm dishes (2 x 10 6 cells/dish) were transfected by calcium phosphate precipitation for 16 hours (Gatz, C, Kaiser, A. & Wendenburg, R. , 1991,Mol. Gen. Genet. 227, 229-237) with the following DNAs: 10 ⁇ g of ZHWTx12-CMV-hGH, 1 ⁇ g pCGNN-ZFHD1-3FKBP, 2 ⁇ g pCGNN- lFRB-p65(361-550) and 7 ⁇ g pUC118.
- Transfected cells were rinsed 2 times with phosphate buffered saline (PBS) and given fresh medium for 5 hours.
- PBS phosphate buffered saline
- cells were removed from the dish in Hepes Buffered Saline Solution containing 10 mM EDTA, washed with PBS/0.1% BSA/0.1% glucose and resuspended in the same at a concentration of 2 x 10 7 cells/ml.
- This reporter gene containing 12 tandem copies of a ZFHD1 binding site
- This reporter gene is identical to pZHWTxl2-CMV-SEAP except the Xbal-Hindlll fragment containing the minimal CMV promoter was replaced with the following Xbal- Hindlll fragment containing a minimal IL2 gene promoter (-72 to +45 with respect to the start site; Siebenlist et al., MCB 3:149, 1986):
- pLH which contains the hygromycin B resistance gene driven by the Moloney murine leukemia virus LTR and a unique internal Clal site
- the hph gene was cloned as a Hindlll-Clal fragment from pBabe Hygro (Morganstern and Land, NAR 18:3587-96,
- a Clal-BstBI fragment consisting of the following was inserted into the Clal site of pLH such that the directions of transcription from the viral LTR and the internal Gal4-IL2 promoters were the same:
- a bicistronic expression vector that directs the production of both ZFHD1-3FKBP and lFRB-p65 through the use of an internal ribosome entry sequence (IRES).
- IRES internal ribosome entry sequence
- This expression plasmid was cotransfected, together with a zeocin-resistance marker plasmid, into a cell line earning a retrovirally- transduced SEAP reporter gene, and a pool of approximately fifty drug-resistant clones was selected and expanded.
- Fig. 4D shows that this pool of clones also exhibited rapamycin-dependent SEAP production with no detectable background and a very similar dose-response curve to that observed in transiently transfected cells.
- rapamycin-responsive gene expression can be readily obtained in both transiently and stably transfected cells. In both cases, regulation is characterized by very low background and high induction ratios.
- Helper-free retroviruses containing the reporter gene or DNA binding domain fusion were generated by transient co-transfection of 293T cells (Pear, W.S., Nolan, G.P., Scott, M.L. & Baltimore D., 1993, Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA 90, 8392-8396) with a
- HT1080 cells infected with retroviral stock were diluted and selected in the presence of 300 ⁇ g/ml Hygromycin B. Individual clones from this and other cell lines described below were screened by transient transfection of the missing components followed by the addition of rapamycin as described above. All 12 clones analyzed were inducible and had little or no basal activity. The most responsive clone, HT1080L, was selected for further study.
- HT20-6 cells which contain the pLH-ZHWTx12-IL2-SEAP reporter gene, ZFHD1-3FKBP DNA binding domain and lFRB-p65(361-550) activation domain stably integrated, were generated by first infecting HT1080L cells with SMTN-ZFHD1-3FKBP- packaged retrovirus and selecting in medium containing 500 ⁇ g/ml G418. A strongly responsive clone, HT1080L3, was then transfected with linearized pCGNN-lFRB- p65(361-550) and pZeoSV (Invitrogen) and selected in medium containing 250 ⁇ g/ml Zeocin.
- HT23 cells were generated by co-transfecting HT1080L cells with linearized pCGNN-1FRB-p65(361-550)-IRES-ZFHD1-3FKBP and pZeoSV and selecting in medium containing 250 ⁇ g/ml Zeocin. Approximately 50 clones were pooled for analysis.
- cells were plated in 96-well dishes (1.5 x 10 4 cells/well) and 200 ⁇ l medium containing the indicated amounts of rapamycin (or vehicle) was added to each well. After 18 hours, medium was removed and assayed for SEAP activity. In some cases, medium was diluted before analysis and relative SEAP units obtained multiplied by the fold-dilution. Background SEAP activity, measured from untransfected HT1080 cells, was subtracted from each value.
- mice Animals, husbandry, and general procedures. Male nu/nu mice were obtained from Charles River Laboratories (Wilmington, MA) and allowed to acclimate for five days prior to experimentation. They were housed under sterile conditions, were allowed free access to sterile food and sterile water throughout the entire experiment, and were handled with sterile techniques throughout. No
- immunocompromised animal demonstrated outward infection or appeared ill as a result of housing, husbandry techniques, or experimental techniques.
- mice To transplant transiently transfected cells into mice, 2 x 10 6 transfected HT1080 cells, were suspended in 100 ⁇ l PBS/0.1% BSA/0.1% glucose buffer, and administered into four intramuscular sites (approximately 25 ⁇ l per site) on the haunches and flanks of the animals. Control mice received equivalent volume injections of buffer alone.
- Rapamycin was formulated for in vivo administration by dissolution in equal parts of N,N-dimethylacetamide and a 9:1 (v:v) mixture of polyethylene glycol (average molecular weight of 400) and polyoxyethylene sorbitan monooleate. Concentrations of rapamycin, in the completed formulation, were sufficient to allow for in vivo
- mice bearing no transfected HT1080 cells, received 10.0 mg/kg rapamycin.
- other control mice bearing transfected cells, received only the rapamycin vehicle.
- mice Blood was collected by either anesthetizing or sacrificing mice via CO2 inhalation. Anesthetized mice were used to collect 100 ⁇ l of blood by cardiac puncture. The mice were revived and allowed to recover for subsequent blood collections. Sacrificed mice were immediately exsanguinated. Blood samples were allowed to clot for 24 hours, at 4°C, and sera were collected following centrifugation at 1000 x g for 15 minutes. Serum hGH was measured by the Boehringer Mannheim non-isotopic sandwich ELISA (Cat No. 1 585 878). The assay had a lower detection limit of 0.0125 ng/ml and a dynamic range that extended to 0.4 ng/ml. Recommended assay instructions were followed.
- rapamycin was administered to mice approximately one hour following injection of HT1080 cells. Rapamycin doses were either 0.01, 0.03, 0.1, 0.3, 1.0, 3.0, or 10.0 mg/kg. Seventeen hours following rapamycin administration, the mice were sacrificed for blood collection.
- mice received 10.0 mg/kg of rapamycin one hour following injection of the cells. Mice were sacrificed at 4, 8, 17, 24, and 42 hours following rapamycin administration.
- mice were administered transfected HT1080 cells as described above. Approximately one hour following injection of the cells, mice received the first of five intravenous 10.0 mg/kg doses of rapamycin. The four remaining doses were given under anesthesia, immediately subsequent to blood collection, at 16, 32, 48, and 64 hours. Additional blood collections were also performed at 72, 80, 88, and 96 hours following the first rapamycin dose. Control mice were administered cells, but received only vehicle at the various times of administration of rapamycin. Experimental animals and their control counterparts were each assigned to one of two groups. Each of the two experimental groups and two control groups received identical drug or vehicle treatments, respectively. The groups differed in that blood collection times were alternated between the two groups to reduce the frequency of blood collection for each animal.
- hGH concentrations in the rapamycin-treated animals compared favorably with normal circulating levels in humans (0.2-0.3 ng/ml). No plateau in hGH production was observed in these experiments, suggesting that the maximal capacity of the transfected cells for hGH production was not reached.
- Control animals those that received transfected cells but no rapamycin and those that received rapamycin but no cells— exhibited no detectable serum hGH. Thus, the production of hGH in these animals was absolutely dependent upon the presence of both engineered cells and rapamycin.
- This half-life is several hundredfold longer than the half-life of hGH itself and approximately twice the half-life of rapamycin (4.6 hr) in these animals.
- the slower decay of serum hGH relative to rapamycin could reflect the presence of higher tissue concentrations of rapamycin in the vicinity of the implanted cells.
- persistence of hGH production from the engineered cells may be enhanced by the stability of hGH mRNA.
- rapamycin concentration is predicted to approach a steady-state trough concentration of 1.7 ⁇ g/ml after two doses (shown as dotted line in Fig. 8).
- hGH levels should also approach a steady state trough concentration following the second dose.
- Fig. 8 shows that treated animals indeed held relatively stable levels of circulating hGH in response to repeated doses of rapamycin. After the final dose, hGH levels remained constant for 16 hours and then declined with a similar half-life as rapamycin (6.8 hours for hGH versus 4.6 hours for rapamycin).
- the use of a small-molecule drug to link a DNA-binding domain and activation domain is an effective strategy for regulating gene expression in vivo.
- One especially attractive feature is that the system is entirely modular, allowing each component to be optimized and engineered independently.
- the dimerization strategy can be adapted to virtually any DNA-binding and activation domain.
- We have used here a DNA-binding domain of defined structure which readily supports rational engineering of DNA-binding affinity and new recognition specificities.
- activation domains can be engineered for maximal potency and other suitable properties.
- engineered transcription factors used in these experiments elicit very high levels of gene expression relative to conventional promoter /enhancer systems, and further enhancements in either domain can be readily incorporated.
- the ability to introduce engineered transcription factors dedicated to the transcription of a single target gene provides opportunities to achieve lower backgrounds and substantially higher levels of gene expression in vivo than conventional expression vectors.
- each individual component of our transcription factor fusion proteins is human in sequence, each protein contains junction peptides which could potentially be recognized as foreign. These junctions may be designed or selected, however, to minimize their presentation to the immune system, as discussed previously.
- rapamycin-based systems The principal limitation of rapamycin-based systems is the native biological activity of rapamycin, which, through inhibition of FRAP activity blocks cell-cycle progression leading to immunosuppression in vivo.
- a therapeutic protein such as hGH
- sub-immunosuppressive doses of rapamycin less than 1 mg/kg.
- Rapamycin purification Rapamycin was obtained by fermentation.
- the rapamycin producing organism Streptomyces hygroscopicus (ATCC# 29253), was cultivated on a complex media in 15 L or 30 L fed-batch fermentations.
- the biomass was harvested after 9-14 days by centrifugation.
- the supernatant was contacted for 1-2 hours with a nonionic, polymeric adsorbent resin, XAD-16 (Rohm and Haas).
- the adsorbent was recovered by centrifugation, combined with the biomass, and extracted repeatedly with methylene chloride. The solvent was removed in vacuo and the resulting residue extracted with acetonitrile which was then condensed in a similar manner.
- Rapamycin obtained exhibited identical HPLC, spectroscopic, and biological characteristics as an authentic sample of rapamycin.
- Rapamycin E and Z-24-(O-ethyloxime) (AP1688 and 1689)
- Rapamycin E and Z
- -24-(O-isobutyloxime) AP1684 and 1685
- Rapamycin E and Z)-24-(O-benzyloxime) (AP1682 and 1683)
- Rapamycin E and Z-24-(O-carboxymethyloxime) (AP1686 and 1687)
- Rapamycin E and Z
- -24-(O-carboxamidomethyloxime) AP1729 and 1730
- Affinities of rapamycin C24 analogs for FKBP were determined using a competitive assay based on fluorescence polarization (FP).
- FP fluorescence polarization
- a fluorescein-labelled FK506 probe (AP1491) was synthesized, and the increase in the polarization of its fluorescence used as a direct readout of % bound probe in an equilibrium binding experiment containing sub-saturating FKBP and variable amounts of rapamycin analog as competitor.
- terr-Butyldimethylsilyl trifluoromethanesulfonate (108 ⁇ L, 470 ⁇ mol) was added dropwise to a stirred solution of FK506 (103 mg, 128 ⁇ mol) and 2,6-lutidine (89.5 ⁇ L, 768 ⁇ mol) in dichloromethane (3 mL) at 0°C. The resulting solution was stirred at 0°C for 2 h, and then treated with MeOH (0.5 mL) and ether (15 mL). The mixture was washed with 10% aqueous NaHCO 3 (3 mL) and brine (3 mL).
- FP buffer prepared using only low-fluorescence reagents from Panvera
- FP buffer bovine gamma globulin
- a probe solution was prepared containing 10 nM AP1491 in 0.1% ethanol/FP buffer, and 100 ⁇ l added to each well with mixing.
- Duplicate control wells contained ethanol instead of rapamycin analog (for 100% probe binding) or ethanol instead of rapamycin analog and FP buffer instead of FKBP (0% binding).
- the plates was stored covered in the dark for approximately 30 min to permit equilibration and then the fluorescence polarization of the sample in each well read on a Jolley FPM-2 FP plate reader (Jolley Consulting and Research, Inc., Grayslake, IL) in accordance with the manufacturer's recommendations.
- the mean polarization (mP units) for each competitor concentration was usually converted to % total binding by reference to the control values and plotted (y) vs. log molar final concentration of competitor (x).
- IC50 was determined by interpolation. Rapamycin and C14-desoxo-rapamycin were included as controls in each case ( C14- desoxo-rapamycin was prepared as described by Luengo, J.I. et al. 1994 Tetrahedron Lett. 35, 6469-6472).
- C7 rapalogs' A series of rapalogs with bulky substituents at C7 (C7 rapalogs') was synthesized using chemistry broadly as described (Luengo et al. 1995. Chemistry and Biology 2, 471-481). The data here refer to the three rapalogs shown below:
- Trifluoroacetic acid (0.084 mL, 1.1 mmol) was added to a stirring solution of rapamycin (50 mg, 0.055 mmol) and indole (64 mg, 0.55 mmol) in 2 mL of dichloromethane at -40°C. The temperature was maintained between -40°C and -45°C for 3 h, then the reaction mixture was partitioned between ethyl acetate (10 mL) and brine (20 mL). The organic phase was then washed with additional brine (4 X 20 mL) and dried over anhydrous sodium sulfate.
- Reverse phase HPLC Reverse phase HPLC (Rainin C-18 ODS 1" column, 65%
- HT20-6 cells which contain a ZFHDl-responsive SEAP reporter and stably express ZFHDl-(3xFKBP) and FRB-p65: see Example 3(B)).
- HT20-6 cells were plated in 96-well dishes (1 x 10 4 cells/well) and 200 ⁇ l medium containing serial dilutions of rapalog was added to each well (in triplicate). After 22 hours, medium was removed and assayed for SEAP activity as described in example 2.
- C7 rapalogs are also assayed for their ability to reduce the proliferation of activated human T cells or murine splenocytes as a measure of their reduced ability to binding FRAP.
- Suitable assays are well known in the art- for example, inhibition of 3 H-thymidine uptake (Luengo et al. 1995. Chemistry and Biology 2, 471-481).
- Example7 Mutagenesis and phage display to generate modified Ligand-Binding Domains complementary to various rapalogs
- Yeast and Candida FRBs modified by analogy to the modified hFRAP FRB domains discussed herein, may also be prepared by substitution of a codon for a different amino acid in place of one or more of the two conserved Phe residues and the conserved Asp and Asn residues within each of their FRB domains.
- Illustrative modified FRB domains derived from TOR 1 and TOR2 include the following:
- An expression vector based on pET20b (Novagen) was constructed using standard procedures that expresses FKBP preceded by a hexahistidine tag and a portion of the H. influenza hemaglutinin protein that is an epitope for the monoclonal antibody 12CA5.
- the sequence of the protein encoded by this vector is as follows:
- oligonucleotide-mediated site-directed mutagenesis was performed on the single- stranded form of the vector prepared from E.coli CJ236, as described (Kunkel, T.A., Bebenek, K. and McClary, J. 1991. Meth Enzymol 204, 235-139). Mutants were confirmed by dideoxy sequencing. Mutant proteins were expressed in E.coli BL21(DE3) (Novagen) as described (Wiederrecht, G. et al. 1992. J. Biol. Chem. 267, 21753-21760), and purified to homogeneity as described (Cardenas, M.E. et al. 1994. EMBO J. 13, 5944-5957).
- mutant human FKBP12 proteins were generated, using the indicated oligonucleotide primers (mutated bases in upper case; 5'->3'):
- FP fluorescence polarization
- a NcoI-BamHI fragment encoding residues 2021-2113 (inclusive) of human FRAP was generated by PCR with primers 28 and 29 (below),and cloned into a derivative of pET20b(+) (Novagen) in which the Ndel site is mutated to Ncol, to create pET- FRAP(2021-2113).
- Single-stranded DNA of this vector was used as a template in site- directed mutagenesis procedures, as described above, to generate vectors encoding FRAPs mutated at rapamycin contact residues. Mutants were confirmed by dideoxy sequencing.
- Mutants were then amplified by PCR using primers (30 and 31) that append Xbal and Spel sites, and cloned into Xbal-Spel digested pCGNN-FRB-p65(361-550) (example 1) to generate a series of constructs directing mammalian expression of chimeric proteins of the form E-N-mutant FRAP(2021-2113)-p65(361-550), where E indicates HA epitope tag and N indicates nuclear localization sequence. Constructs were verified by restriction digestion and dideoxy sequencing.
- mutant FRAPs that potentially bind to C7 rapalogs were generated using the indicated oligonucleotide primers (mutated bases in upper case; 5'- >3'):
- each construct is transiently co-transfected into human HT1080B14 cells, as described in example 2. Following transfection, serial dilutions of rapamycin or rapalog are added to the culture medium. After 24 hours, SEAP activity is measured as described in example 2; the potency of SEAP activation at various rapalog concentrations is proportional to the affinity of the FRAP mutant for the complex between FKBP and the rapalog.
- a derivative of pCANTAB-5E was constructed by site-directed mutagenesis with primer 1 in which Cys202 of fd genelll is replaced with Tyr. Cys202 can disrupt display of proteins fused at Aspl98 due to the formation of mixed disulfides, a problem alleviated by the Tyr substitution (Cunningham, B.C. et al. 1994. EMBO J. 13, 2508). The construct was verified by DNA sequencing and then digested with Ncol and BamHI.
- oligonucleotides (2 and 3) was ligated in to yield the plasmid pCANTAB-AP-poly, which contains a NcoI-Spel-BamHI polylinker between the Ncol site in the genelll signal sequence and the BamHI site at codon 198 in genelll.
- This vector can accept in frame
- NcoI-BamHI fragments to give constructs directing the N-terminal display of the encoded protein on pill residues 198-406, the fusion configuration described by Cunningham et al. (1994. EMBO J. 13, 2508).
- a fragment of FRAP encoding residues 2015-2114 inclusive was amplified from pCGNN-FRAPi by PCR with Pfu polymerase (Stiratagene) and the primers 4 and 5. The fragment was purified, digested with Ncol and BamHI, and ligated into NcoI-BamHI digested pCANTAB-AP-poly to yield the vector p-CANTAB-FRAP(2015-2114), which was verified by DNA sequencing. (b) Preparation of His6-flag-FKBP
- an expression vector based on pET20b (Novagen) was constructed using standard procedures that expressed FKBP preceded by a hexahistidine tag (for affinity purification) and 'flag' sequence (Kodak IBI) (for immunological detection).
- the protein was expressed in E.coli BL21(DE3 (Novagen) as described (Wiederrecht, G. et al. 1992. J. Biol. Chem. 267, 21753-21760) and purified to homogeneity using Ni-NTA agarose (Qiagen) as described (Cardenas, M.E. et al. 1994. EMBO J. 13, 5944-5957).
- the sequence of the expressed protein was as follows:
- p-CANTAB-AP-FRAP(2015-2114) was transformed into E.coli XL-1 (Stiratagene) and display phage prepared by rescue with helper phage K07 essentially as described (Lowman, H.B, and Wells, J.A. Methods: Comp. Methods Enzymol. 1991. 3, 205-216) except that the overnight culture used to innoculate the phagemid growth was grown in medium containing 2% glucose, and the phage growth was carried out at 25°C after an initial 1 hour incubation at 37°C to allow K07 infection.
- phagemid particles were also prepared from cells transformed with the chloramphenicol- resistant vector pBC (Stratagene). Phagemid titers (colony-forming units, cfu) in each case were determined as described (Lowman, H.B, and Wells, J.A. Methods: Comp.
- Carbenicillin (carb) and chloramphenicol (chlor) -resistant cfu titers were determined as described above, and the carb/chlor ratio used a a measure of the specific enrichment of (carbenicillin-resistant) FRAP phagemids.
- the results showed a dramatic enrichment (271 -fold) of FRAP phage compared to the starting mixture after affinity enrichment on the rapamycin-FKBP matrix, but not on the FK506-FKBP matrix (2.4-fold) or FKBP alone (1.5-fold).
- This experiment demonstrates the procedure that is used to purify from a library mutant FRAPs that bind rapamycin analogs bearing bumps on the FRAP binding side of the molecule (for example at the C7 position).
- the coding sequence of FKBP (amino acids 1-107) was amplified by PCR with primers 1 and 2, using as template 5 ng of plasmid pCGNN-F1 (US Patent Application Serial No. 08/581,713 (filed December 29, 1995)).
- the PCR product was purified, digested with Ncol and BamHI and ligated into NcoI-BamHI digested pCANTAB-AP- poly to yield the vector pCANTAB-AP-FKBP which was verified by restriction analysis and DNA sequencing.
- This construct directs the N-terminal display of FKBP on pill residues 198-406 where Cys202 has been mutated to Tyr: the configuration described by Cunningham et al. (1994. EMBO J. 13. 2508).
- pCANTAB-AP-FKBP was transformed into E.coli XL-1 (Stiratagene) and display phage prepared by rescue with helper phage K07, essentially as described (Lowman, H. B. and Wells, J.A. Methods: Comp. Methods Enzymol. 1991, 3, 205-216) except that the overnight culture used to innoculate phagemid growth was grown in medium containing 2% glucose. Phagemid titers (colony-forming units, cfu) were determined as
- this experiment demonstrated specific, high-affinity binding of the displayed FKBP for FK506.
- the IC50 for the interaction in this assay was 0.65 nM, in excellent agreement with the published Kd of FKBP for FK506 of 0.4-1 nM.
- This experiment established that display of FKBP on phage could be used as a means to affinity select, from a library of mutants, FKBPs that have affinity for bumped ligands.
- the degenerate primers 3 and 4 were used to reamplify a portion of the FKBP gene and introduce the degenerate codon NNS (where N is any base and S is G or C) in place of the codons for Tyr26, Phe36, Asp37, Arg42 and Phe99.
- NNS any base and S is G or C
- the sidechains of these residues were observed using computer graphics to be contact with the C14 carbonyl and/or C13 hydroxyl, or to abut residues that are in contact, and were therefore candidates for mutation to generate mutants that might have good affinity for analogs with bumps introduced at these positions.
- Convenient ApaLI and BamHI sites that flank the randomized region and are unique in pCANTAB-AP- FKBP were used.
- FKBP FKBP
- the PCR product was purified using the QIAEX II kit (Qiagen), digested extensively with ApaLI and BamHI, and ligated into ApaLI-BamHI digested pCANTAB- AP-FKBP.
- Ligation products are purified by phenol extraction, concentrated by ethanol precipitation and electroporated by standard techniques (Dower, W.J. et al. 1988. Nucleic Acids Res. 16, 6127) into E.coli XL-1 cells. Phage are rescued and titered as described previously (Lowman, H. B. and Wells, J.A. Methods: Comp. Methods Enzymol 1991, 3, 205-216).
- cfu phagemid particles are prepared in 250 ⁇ l PBS/3% BSA/0.05% tween-20 (PBSBT).
- Recombinant GST-FRAP fusion protein (expressed in E.coli and purified as described by Chen et al. 1995. Proc. Natl. Acad. Sci. USA. 92, 4947- 4951) is added to a final concentration of 1 ⁇ M, followed by 2.5 ⁇ l of a 10 ⁇ M stock of bumped rapamycin 100% ethanol. The mixture is incubated for 1 hour at room
- Bound phagemids are eluted from the beads by adding 200 ⁇ l 0.2 M glycine pH 2.0 and incubating for 10 min. The beads are pelleted and the supernatant (containing eluted phagemids) collected and neutralized with 26 ⁇ l 1 M Tris base.
- Carbenicillin (carb) and chloramphenicol (chlor) -resistant cfu titers are determined as described above, and the carb/chlor ratio used as a measure of the specific enrichment of (carbenicillin-resistant) FRAP phagemids.
- FKBP phage clones Titers of phage are monitored over several rounds of selection, and when specific enrichment over background is detected, individual FKBP phage clones can be isolated and sequenced by standard techniques. Binding of the clones to bumped rapamycins is measured by competitive ELISA or fluorescence polarization assay.
- Rapamycin and its analogs may be used to trigger activation of a receptor effector domain by oligomerizing chimeric proteins, one of which contains one or more FKBPs and an effector domain and the other of which contains one or more FRAP domains and an effector domain. This scheme is illustrated in Figure T1(a). While both proteins are shown anchored to the membrane, a single one could be membrane anchored, and addition of rapamycin or analog would recruit the second protein to the membrane via dimerization.
- Membrane anchoring may be effected through a transmembrane protein anchor or through lipid modification of the protein(s), such as myristoylation.
- the same effector domain may be present on both proteins, or different protein domains that interact functionally may be used, such as a protein kinase and a protein kinase substrate.
- a second effector may serve to inhibit the activity of the first effector.
- the chimeric proteins are mixed chimeras, discussed previously, and contain FKBP and FRAP domains together with the heterologous effector domain. Oligomerization of a single mixed chimera may also be used to activate signal transduction, as shown in Figure T1(b). Here rapamycin is shown to dimerize two identical copies of the protein. Reiteration of the FKBP and FRAP domains permits higher multiples to occur, subject to geometric constraints.
- rapamycin in signal transduction is to trigger receptor tyrosine kinase activation and to trigger apoptosis via Fas activation, both of which are discussed below.
- DNA manipulations were performed following standard procedures (F.M. Ausubel et al, Eds., Current Protocols in Molecular Biology, John Wiley & Sons, New York, 1994) and all protein protocols were performed following standard procedures (Harlow, E. and Lane, D. 1988. Antibodies, a Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor.). All PCR products used to make constructs were confirmed by sequencing.
- Rapamycin-inducible receptor tyrosine kinase activation A. Rapamycin-inducible receptor tyrosine kinase activation.
- a Xbal-Myr-BamHI cassette obtained by annealing oligonucleotides 1 and 2, was digested with Xbal /BamHI and cloned into the Xbal /BamHI site of the pCG expression vector (Tanaka, M. and Herr, W. 1990. Cell 60: 375-386) to create pCGM. (For oligonucleotide sequences, see (7) below).
- This oligonucleotide cassette consists of an inframe Xbal site followed by sequence encoding for the first 15 amino acids residues of c-Src tyrosine kinase that has been shown to allow myristoylation and target protein to the plasma membrane (Cross et al., 1984. MCB. 4:1834-1842).
- the myristoylation domain is followed by an inframe Spel site and stop codons.
- the Xbal site in the pCG vector is placed such that it adds two amino acids between the initiating Met and the sequence cloned. Since the spacing between the initiating Met and the myristylated Gly is crucial for membrane localization of c-Src (Pellman et al 1985. PNAS.
- the Xbal site following the ATG in pCGM was deleted by site directed mutagenesis following manufacturers protocol (Muta-Gene, BioRad). To facilitate future cloning steps the Spel site in the myristylation cassette was mutated to a Xbal site.
- Single stranded uracil-DNA of pCGM was prepared and the mutagenesis was carried out using both oligonucleotide 3 (to delete the Xbal site following ATG and add an EcoRI site 5' to ATG) and oligonucleotide 4 (to change the Spel site following the myristylation domain to a Xbal site).
- the resulting sequence surrounding the ATG of the pCM vector was confirmed by sequencing using oligonucleotide 5 (see sequence 1, (8) below).
- a Spel-HA-BamHI cassette was prepared by annealing complementary oligonucleotides (oligonucleotides 6 and 7). This cassette has an inframe Spel site followed by nine amino acids of H. influenzae hemaglutinin gene that is recognized by the monoclonal antibody 12CA5, stop codons and a BamHI site.
- the Spel-HA-BamHI cassette was sub cloned into the Spel/BamHI site of pCGNNFl, pCGNNF2 and pCGNNF3. Subsequently, the 1/2/3 copies of FKBP fused with HA epitope was sub cloned as an Xbal/BamHl fragment into pCM.
- the resulting plasmid (pCMF1/2/3.HA) has the following features: myristylation domain; an inframe Xbal site; one/two/three copies of FKBP; an inframe Spel site; a HA epitope tag; and stop codons.
- a Spel-Flag-BamHI cassette can be prepared by annealing complementary oligonucleotides (oligonucleotides 8 and 9). This cassette has the same features as the Spel-HA-BamHI cassette described above with the exception that the inframe Spel site is followed by sequence that codes for eight amino acids (DYKDDDDY) (Hopp et al., 1988. Biotech. 6: 1205-1210) that is recognized by a monoclonal antibody anti-FLAG.M2 (Kodak Scientific Imaging Systems).
- the Spel-Flag-BamHI cassette is sub cloned into the Spel/BamHI site in pCGNN-1FRB, pCGNN-2FRB and, pCGNN-3FRB.
- FRB domain-Flag epitope fusions are sub cloned as a Xbal/BamHI fragment into pCM.
- the resulting plasmid (pCMFR1/2/3.Flag) has the following features: myristylation domain; an inframe Xbal site; one/two/three copies of FRB; an inframe Spel site; a Flag epitope tag; and stop codons. 4. Fusion of FKBP and FRB constructs to receptor tyrosine kinase cytoplasmic domain
- the cytoplasmic domain of receptor tyrosine kinase of choice (e.g., EGFR, erbB-2, PDGFR, KDR/Flk-1, Flt-1) is PCR amplified with inframe 5'XbaI and 3' Spel sites.
- the PCR product may be subcloned either into the inframe Xbal site such that the Xbal site is restored, or into the inframe Spel site such that the Spel site is restored in pCMFR series or pCMFseries vectors (see above).
- the FKBP/FRB domain(s) can be placed either C-terminal or NH2-terminal to the cytoplasmic domain of the receptor tyrosine kinase.
- the vectors are constructed such that (i) the cytoplasmic domain of a given receptor is fused to both FKBP and FRB (for e.g., EGFR cytoplasmic domain fused to either FKBP or FRB) or (ii) can be constructed such that cytoplasmic domains of two different receptors are fused to FKBP and FRB (for e.g., EGFR cytoplasmic domain fused to FKBP and erbB-2 cytoplasmic domain fused to FRB).
- the constructs of choice e.g., pCMEGFR-FR1 and pCMEGFR-F1 are cotransfected into Cos-1 cells by lipofection (Gibco BRL).
- lysis buffer 1% Triton X-100; 50mM Tris.cl pH8.0; 150mM NaCl; 5mM NaF; lmM sodium ortho vanadate; 10ug/ml aprotinin; 10ug/ml leupeptin).
- fusion proteins from rapamycin- treated and untreated cell lysates are immunoprecipitated with anti-Flag and 12CA5 antibodies and immunoblotted with anti-phosphotyrosine antibody.
- the choice of cell type; the amount of DNA transfected; the concentration of rapamycin used and the duration of drug treatment are varied to achieve optimal results.
- a selected mammalian cell line (e.g., NIH3T3) is cotransfected with constructs encoding for FRB and FKBP fusion proteins (e.g., pCMEGFR-FR1 and pCMEGFR-F1) and stable cell lines expressing the fusion proteins are established.
- FRB and FKBP fusion proteins e.g., pCMEGFR-FR1 and pCMEGFR-F1
- stable cell lines expressing the fusion proteins are grown either in the presence or absence of rapamycin and the changes in cell growth rate are determined by routine procedures (e.g., by monitoring cell number; by determining the 3 H thymidine incorporation rate, etc.).
- the choice of receptor tyrosine kinase; the type of receptor activation (homodimer vs. heterodimer) may be chosen to obtain optimal results.
- the ability to control Fas activation and trigger apoptosis via a small molecule has applications both in gene therapy, where it may be used to selectively eliminate engineered cells, and in experimental systems.
- the proteins described here are anchored to the membrane via the low affinity NGF receptor, also called p75. It should be appreciated, however, that another protein anchor could be readily substituted.
- p75 is useful experimentally because of the availability of antibodies to its extracellular domain, and its lack of high affinity interaction with any identified ligand (Bothwell, M. 1995. Annu. Rev. Neurosci. 18:223-253).
- Vectors to direct the expression of FRAP-Fas fusion proteins containing the extracellular and transmembrane domain of the low affinity NGF receptor were derived from the mammalian expression vector pJ7W (Morgenstern, J.P. and Land, H. 1990. Nucleic Acids Res. 18:1068), modified by substitution of a pUC backbone for the original pBR backbone using standard methods. We call this vector pATVV. Inserts cloned into the polylinker sites of this plasmid are transcribed under the control of the simian CMV promoter and enhancer sequences.
- the polylinker follows the CMV sequence with Hind III-Sall-Xbal-BamHI-Smal-Sstl-EcoRI-Clal-KpnI-BgIII. Any mammalian expression vector with suitable cloning sites and promoter could be substituted.
- a restriction fragment encoding a fragment of p75 flanked by Hindlll and Xbal sites was generated by PCR using primers J1 (5') and J2 (3'), based on the sequence of p75 (Johnson, D., Lanahan, A., Buck, C.R., Shegal, A., Morgan, C, Mercer, E., Bothwell, M., Chao, M. 1986. Cell 47:545-554).
- the original source of the PCR template was a clone derived from a human brain library, using primers similar to J1 and J2 but with different restriction sites.
- the 5' end of the resulting fragment contains a Hindlll site followed by an EcoRI site, a Kozak sequence and the initiation of p75 coding sequence (amino acid 1).
- the 3' end generated encodes the receptor sequence up to and including amino acid 274, 2 amino acids past the predicted membrane spanning sequence, followed by an Xbal site. Analogous portions of other transmembrane receptors can be substituted for this fragment.
- the PCR product was subcloned as a Hindlll-Xbal fragment into Hindlll-Xbal cut pA7W, generating pA7Wp75. The construct was verified by restriction analysis and DNA sequencing.
- Fas amino acids 206-304 Fas amino acids 206-304
- Fas amino acids 206-319 FasL
- the primers used were J3 (5') and J4 or J5 (3'). J5 generates a fragment of Fas that ends beyond its termination codon; when cut with Spel, the nucleotides encoding the terminal 15 aa of Fas are removed to give a truncated form of intracellular Fas we call Fass. Removal of these 15 aa increases the activity of Fas in some cell types (Itoh, N., and Nagata, S. 1993. J. Biol.
- influenza haemagglutinin protein (E) 3' to the Spel site, followed by a BamHI site. Cutting the resultant plasmid with Xbal and BamHI generated fragments encoding Fas followed by the epitope tag (designated E for these constructs).
- E epitope tag
- p75-FRAP-Fas-epitope fusion proteins addition of FRAP-containing fragments to pA7Wp75-Fas S E and pA7Wp75-Fas L E to generate p75-FRAP x -Fas SorL E and p75- Fas SorL -FRAP x E
- Xbal-Spel fragments containing a portion of FRAP are described previously in this document. These Xbal-Spel fragments were inserted into either the Xbal site directly after the p75 coding sequence to generate p75-FRAP x -Fas SorL E or in to the Spel site directly after the Fas fragment to generate p75-Fas SorL -FRAP x E. Alternatively, more than one FRAP fragment is subcloned in, either as a FRAPn fragment, or by sequential subcloning of Xbal-Spel fragments into the Spe I site available after subcloning the first FRAP into either Xbal or Spel.
- the final series of vectors encodes (from the N to the C terminus) p75 extracellular and transmembrane sequence, one or more FRAP-derived domains fused N- or C-terminally to one or more Fas intracellular domains, and an epitope tag.
- the Xbal-Spel fragments containing one or more FKBPs have been described elsewhere in this document. These fragments were inserted into either the Xbal site directly after the p75 coding sequence to generate p75-FKBP n -Fas SorL or into the Spel site directly after the Fas fragment to generate p75-Fas SorL -FKBP n .
- the final series of vectors encodes (from the N- to the C-terminus) p75 extracellular and transmembrane sequence, one or more FKBPs fused N- or C-terminally to one or more Fas intracellular domains, and an epitope tag.
- transient transfections the two plasmids to be tested are cotransfected into a cell line such as HT1080 by a standard method such as lipofection, calcium phosphate precipitation or electroporation.
- a cell line such as HT1080
- lipofection, calcium phosphate precipitation or electroporation One or more days after transfection, cells are treated with no addition or one or more concentrations of rapamycin or one or more
- FK1012 serves as a positive control that the FKBP-Fas construct is functional.
- FK1012 serves as a positive control that the FKBP-Fas construct is functional.
- Several hours to 1 day later, the cells are monitored for response by one of several methods.
- Cell lysates were prepared by conventional means and used to generate Western blots that are probed with antibody directed against HA or against the extracellular domain of p75.
- cells can be assayed by collection in isotonic solution plus 10 mM EDTA, stained with anti-p75 monoclonal antibody and labeled secondary antibody, and the positive cells measured by FACS.
- a decrease in either Western blot signal or FACS signal upon treatment indicates sucessful induction of cell death (or decrease in protein expression).
- commercially available kits can be used to monitor apoptosis.
- a vector encoding a selectable marker such as neomycin resistance is cotransfected along with the plasmids described.
- a selectable marker such as neomycin resistance
- An alternative means of generating stable cell lines expressing the constructs of interest is to subclone the inserts into a retroviral vector.
- the inserts are excisable with Eco RI to facilitate this subcloning.
- the vector is then used to make transducing supernatants by a packaging cell using conventional methods.
- the three dimensional structure of the ternary complex between human FKBP12, rapamycin, and a portion of human FRAP encompassing the minimal FRB domain may be considered.
- Requirements for homodimerization of two molecules of fusion proteins containing FRAP, FKBP and Fas moieties include (i) sufficient length and flexibility of the polypeptide to accomodate the distortions necessary for the FRAP-FKBP interaction to occur between molecules tethered at the membrane, while preserving the ability of aggregated Fas to transduce a signal; and (ii) prevention or minimization of intramolecular dimerization by rapamycin, an event expected to be highly entropically favored due to the chelate effect, and therefore to prevent the desired intermolecular molecular dimerization.
- FRB and FKBP should be joined with a polypeptide linker sufficiently short that intramolecular dimerization is sterically prevented.
- the currently preferred configuration is FRAP-FKBP as the C-terminus of FRAP and the N-terminus of FKBP are distant, allowing a long linker (>ten amino acids) that should still prevent intramolecular dimerization yet afford flexibility.
- This FRAP-FKBP 'cassette' can be present membrane-proximally (i.e. with Fas domain(s) added to the C-terminus), or membrane distal (with the Fas domain membrane-proximal and the FRAP-FKBP cassette appended C-terminally).
- a long linker should be present N-terminal to the FRAP-FKBP domains, to allow for the structural distortions implied by dimerization at the membrane or if the domains are added C-terminally. Again a N-terminal location of FRAP is preferred as this long linker can then comprise natural FRAP sequence from the region N-terminal to the FRB domain, minimizing the immunogenicity of the chimeric protein.
- the twelve fusions were made as Xbal-BamHI cassettes that could be cloned directly as a single fragment, using the three-primer PCR splicing method (Yon, J. and Fried, M. 1989. Nucleic Acids Res. 17, 4895). Cloning in this way avoided the introduction of restriction sites between the genes that would encode foreign sequence and alter the length of the linker.
- a mixture of 1 ng each of pCGNN-lFRAP i and pCANTAB-AP- FKBP was amplified using Pfu polymerase with 1 ⁇ M each of two outer primers (A and C), in the presence of 0.01 ⁇ M of a single 'splice' oligo (B) complementary to both genes that directs the desired fusion.
- the primers used are tabulated below:
- PCR products were purified, digested with Xbal and BamHI, and ligated into Xbal- BamHI digested pCM. The constructs were verified by restriction analysis and DNA sequencing.
- FKBP and FRB are separated by a Fas fragment.
- the starting points for these constructs are pCMF1HA, pCMF2HA, and PCMF3HA.
- FKBP and FRB fragments were cloned into the pCM backbones as Xbal-BamHI fragments that included a Spel site just upstream of the BamHI site. As Xbal and Spel produce compatible ends, this allowed further Xbal-BamHI fragments to be inserted downstream of the initial insert.
- cloning of an Xbal-Spel fragment results in the addition of the fragment at the 5' end of the construct.
- the final p75-anchored construct was made by subcloning the Xbal-Spel fragments shown in Table 1 ((d) below) into pA7Wp75-FassE.
- a similar series is made by subcloning into pA7Wp75-Fas L E. Insertion into vector cut with Xbal resulted in addition of the insert 3' to the p75 fragment. Insertion into this vector cut with Spel resulted in addition of the insert 3' to the Fas fragment.
- Xbal-BamHI fragments from constructs A30 and A31 were cloned into pCM to generate M30 and M31, constructs that direct the expression of MT5Fas ⁇ E and MT ⁇ FassE, where M denotes a myristoylation domain (see this example sections A.1. and A.8.) and other abbreviations are as described in d, table 1.
- EcoRI-BamHI fragments containing these expression cassettes were then cloned into the retroviral vector pSMTN3 (Example 1).
- Helper-free retroviruses containing this DNA were generated by transient co-transfection of 293T cells (Pear, W.S. et al. 1993. Proc. Natl. Acad. Sci. USA, 90, 8392-8396) with the constructs and a Psi(-) amphotropic packaging vector.
- HT1080 cells were infected with viral stock and selected with G418.
- Figure 13 shows that the survival of cells stably transfected with (a) M30 and (b) M31- expressing constructs is potently reduced in the presence of rapamycin, in a dose-dependent manner.
- the extent of cell death is comparable to that of cells expressing a myristoylated (FKBP x 2)-Fas construct (as disclosed in PCT/US94/08008) treated with a synthetic FKBP homodimerizer AP1428:
- AP1428 is compound 49, disclosed in PCT /US95/ 10559. Note that rapamycin has minimal effects on survival of cells not expressing mixed chimeric proteins ( Figure 13(c)).
- transcription factor function at the level of DNA binding is an alternative approach to small molecule control of therapeutic gene expression.
- One approach to creating a transcription factor that is dependent upon rapamycin for DNA binding is to exploit the fact that many transcription factors bind to their cognate sequences as dimers.
- the DNA binding domains of such transcription factors can often be subdivided into a region involved in directly contacting DNA and a region involved in mediating dimer formation.
- FKBP or FRAP the chimeric transcription factor should display rapamycin- dependent DNA binding.
- Any human dimeric transcription factor may be amenable to such an approach.
- Examples include HNF-1, SRF or other MADS box containing proteins, any member of the helix-loop-helix transcription factor family e.g. MyoD, myogenin, any member of the bZIP transcription factor family e.g. FOS, JUN, ATF, CREB.
- rapamycin mediated chimeric protein dimers can bind but naturally occuring parental dimers cannot.
- Lex-FKBP and Lex-FRAP proteins were translated in vitro and mixed together in a binding reaction with radiolabeled LexCon oligonucleotide. Rapamycin was included in binding reactions at the concentrations shown (Lanes 1-4).
- the arrow indicates the position of rapamycin dependent protein-DNA complexes.
- the sequence of the LexCon probe is shown below, the LexA binding site is underlined:
- Plasmids p19BL87, p19BL87G6FKBP and pl9BL87FRB are constructed in pET- 19BHA, a pET-19B based vector modified such that all expressed proteins contain an amino-terminal His.Tag followed by a Haemaglutinin epitope Tag
- p19BL87 encodes the LexA DNA binding domain (aa 1-87).
- LexA coding sequence was amplified by PCR from pCGNNLex202 with primers LexA Xba and LexA87 Spe/Bam. The PCR product was digested with Xbal and BamHI and ligated between the Xbal and BamHI sites of pl9BHA.
- pl9BL87G6FKBP encodes LexA (aa 1-87) fused in frame to FKBP (aa2-108) via a six Glycine flexible linker.
- FKBP coding sequence was amplified by PCR from pCGNNF1 with primers 5'XG6FKBP and FKBP 3' Spe/Bam.
- pCGNNZFHD1-FKBPx3 (ATCC Accession No. 97399) may also be used as a source for FKBP-encoding DNA. See USSN 08/581,713, filed December 29, 1995.
- the PCR product was digested with Xbal and BamHI and ligated between the Spel and BamHI sites of p19BL87.
- pl9BL87FRB encodes LexA (aa 1-87) fused in frame to FRAP (aa2025-2113).
- FRAP coding sequence was isolated as an Xbal-BamHI fragment from pCGNN-GAL4-1FRB and ligated between the Spel and BamHI sites of p19BL87.
- p19BL87G6FKBP and pl9BL87FRB plasmids were translated in vitro using a TNT coupled reticulocyte lysate system (Promega) according to manufacturer's instructions.
- Binding reactions contained 15 ml of binding buffer (10 mM Tris pH 7.5, 1 mM DTT,
- EDTA 0.1 mM EDTA, 10% v/v glycerol, 5 mM MgCl 2 , 60 mM NaCl), 50 mg/ml Bovine Serum Albumin (NEB); 200ng Poly(dIdC)-Poly(dIdC) (Pharmacia); 1ml each of L87G6FKBP and L87FRB programmed reticulocyte lysate and 0.2 ng g-32p-dATP labeled LexCon probe in a total volume of 20 ml. Rapamycin was added to the appropriate concentration and reactions were incubated at room temperature for 30 min.
Abstract
Description






















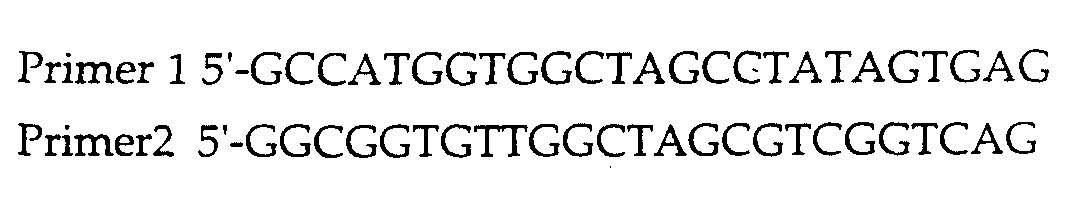


































Claims



Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP96921491A EP0833894B1 (en) | 1995-06-07 | 1996-06-07 | Rapamcycin-based regulation of biological events |
| AU62706/96A AU714904C (en) | 1995-06-07 | 1996-06-07 | Rapamcycin-based regulation of biological events |
| DE69637641T DE69637641D1 (en) | 1995-06-07 | 1996-06-07 | RAPAMYCIN-BASED REGULATION OF BIOLOGICAL ACTIVITIES |
| JP50324497A JP2002514893A (en) | 1995-06-07 | 1996-06-07 | Rapamycin-based regulation of biological events |
| US09/012,097 US6187757B1 (en) | 1995-06-07 | 1998-01-22 | Regulation of biological events using novel compounds |
| US09/781,804 US6649595B2 (en) | 1995-06-07 | 2001-02-12 | Regulation of biological events using novel compounds |
| US10/341,967 US20030206891A1 (en) | 1995-06-07 | 2003-01-14 | Rapamycin-based biological regulation |
| US10/716,062 US20040082515A1 (en) | 1995-06-07 | 2003-11-18 | Regulation of biological events using novel compounds |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US48194195A | 1995-06-07 | 1995-06-07 | |
| US08/481,941 | 1995-06-07 | ||
| US59877696A | 1996-02-09 | 1996-02-09 | |
| US08/598,776 | 1996-02-09 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US48194195A Continuation-In-Part | 1995-06-07 | 1995-06-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1996041865A1 true WO1996041865A1 (en) | 1996-12-27 |
Family
ID=27047093
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1996/009948 WO1996041865A1 (en) | 1995-06-07 | 1996-06-07 | Rapamcycin-based regulation of biological events |
Country Status (3)
| Country | Link |
|---|---|
| KR (1) | KR19990022651A (en) |
| CA (1) | CA2219080A1 (en) |
| WO (1) | WO1996041865A1 (en) |
Cited By (114)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999030164A1 (en) * | 1997-12-09 | 1999-06-17 | President And Fellows Of Harvard College | Method to identify transcriptional modulators |
| WO1999036553A2 (en) * | 1998-01-15 | 1999-07-22 | Ariad Gene Therapeutics, Inc. | Regulation of biological events using multimeric chimeric proteins |
| WO1999058700A1 (en) * | 1998-05-11 | 1999-11-18 | Ariad Gene Therapeutics, Inc. | Multiviral compositions and uses thereof |
| WO2000004178A1 (en) * | 1998-07-14 | 2000-01-27 | Aventis Pharma Deutschland Gmbh | Expression system containing chimeric promoters with binding sites for recombinant transcription factors |
| WO2000023600A1 (en) * | 1998-10-19 | 2000-04-27 | Ariad Gene Therapeutics, Inc. | Materials and methods involving conditional aggregation domains |
| WO2000052179A2 (en) * | 1999-03-03 | 2000-09-08 | Genelabs Technologies, Inc. | Dna binding compound-mediated molecular switch system |
| WO2001000815A1 (en) * | 1999-05-28 | 2001-01-04 | Sangamo Biosciences, Inc. | Molecular switches |
| US6171820B1 (en) | 1995-12-07 | 2001-01-09 | Diversa Corporation | Saturation mutagenesis in directed evolution |
| US6187757B1 (en) | 1995-06-07 | 2001-02-13 | Ariad Pharmaceuticals, Inc. | Regulation of biological events using novel compounds |
| WO2001014387A1 (en) * | 1999-08-24 | 2001-03-01 | Ariad Gene Therapeutics, Inc. | 28-epirapalogs |
| US6238884B1 (en) | 1995-12-07 | 2001-05-29 | Diversa Corporation | End selection in directed evolution |
| US6270998B1 (en) | 1991-04-26 | 2001-08-07 | Osaka Bioscience Institute | DNA coding for human cell surface antigen |
| WO2001068705A2 (en) | 2000-03-16 | 2001-09-20 | Amgen Inc. | Il-17 receptor like molecules and uses thereof |
| US6306649B1 (en) | 1995-06-27 | 2001-10-23 | Ariad Gene Therapeutics, Inc. | Heterologous transcription factors |
| US6352842B1 (en) | 1995-12-07 | 2002-03-05 | Diversa Corporation | Exonucease-mediated gene assembly in directed evolution |
| US6358709B1 (en) | 1995-12-07 | 2002-03-19 | Diversa Corporation | End selection in directed evolution |
| US6361974B1 (en) | 1995-12-07 | 2002-03-26 | Diversa Corporation | Exonuclease-mediated nucleic acid reassembly in directed evolution |
| US6506379B1 (en) | 1995-06-07 | 2003-01-14 | Ariad Gene Therapeutics, Inc. | Intramuscular delivery of recombinant AAV |
| US6537776B1 (en) | 1999-06-14 | 2003-03-25 | Diversa Corporation | Synthetic ligation reassembly in directed evolution |
| WO2003027671A2 (en) * | 2001-09-25 | 2003-04-03 | Eirx Therapeutics Limited | Apoptosis |
| US6562594B1 (en) | 1999-09-29 | 2003-05-13 | Diversa Corporation | Saturation mutagenesis in directed evolution |
| WO2003064383A2 (en) | 2002-02-01 | 2003-08-07 | Ariad Gene Therapeutics, Inc. | Phosphorus-containing compounds & uses thereof |
| US6713281B2 (en) | 1995-12-07 | 2004-03-30 | Diversa Corporation | Directed evolution of thermophilic enzymes |
| US6713279B1 (en) | 1995-12-07 | 2004-03-30 | Diversa Corporation | Non-stochastic generation of genetic vaccines and enzymes |
| US6740506B2 (en) | 1995-12-07 | 2004-05-25 | Diversa Corporation | End selection in directed evolution |
| US6900043B1 (en) | 1999-09-21 | 2005-05-31 | Amgen Inc. | Phosphatases which activate map kinase pathways |
| WO2005078104A1 (en) | 2004-02-09 | 2005-08-25 | Synamem Corporation | Method for generating tethered proteins |
| US6939689B2 (en) | 1995-12-07 | 2005-09-06 | Diversa Corporation | Exonuclease-mediated nucleic acid reassembly in directed evolution |
| US6984635B1 (en) | 1998-02-13 | 2006-01-10 | Board Of Trustees Of The Leland Stanford Jr. University | Dimerizing agents, their production and use |
| US7018793B1 (en) | 1995-12-07 | 2006-03-28 | Diversa Corporation | Combinatorial screening of mixed populations of organisms |
| US7067526B1 (en) | 1999-08-24 | 2006-06-27 | Ariad Gene Therapeutics, Inc. | 28-epirapalogs |
| EP1699415A2 (en) * | 2003-12-29 | 2006-09-13 | Ramot at Tel-Aviv University Ltd. | An assay for the detection of rapamycin and rapamycin analogs |
| US7109317B1 (en) | 1998-11-06 | 2006-09-19 | President And Fellows Of Harvard College | FK506-based regulation of biological events |
| US7189506B1 (en) | 1999-03-03 | 2007-03-13 | Genelabs Technologies, Inc. | DNA binding compound-mediated molecular switch system |
| US7408047B1 (en) | 1999-09-07 | 2008-08-05 | Amgen Inc. | Fibroblast growth factor-like polypeptides |
| US7459540B1 (en) | 1999-09-07 | 2008-12-02 | Amgen Inc. | Fibroblast growth factor-like polypeptides |
| EP2077279A1 (en) | 2000-06-28 | 2009-07-08 | Amgen Inc. | Thymic stromal lymphopoietin receptor molecules and uses thereof |
| US7579180B2 (en) | 1999-03-26 | 2009-08-25 | Amgen Inc. | Beta secretase polypeptides |
| US7629315B2 (en) | 2005-03-09 | 2009-12-08 | University Of Pittsburgh Of The Commonwealth System Of Higher Education | Compositions for blocking the inhibitory effect of human CRP on human leptin |
| EP2130920A2 (en) | 2000-03-28 | 2009-12-09 | Amgen, Inc | Beta-like glycoprotein hormone polypeptide and heterodimer |
| US7662924B2 (en) | 2002-02-22 | 2010-02-16 | The Board Of Trustees Of The University Of Illinois | Beta chain-associated regulator of apoptosis |
| US7833741B2 (en) | 2002-08-07 | 2010-11-16 | Ambit Biosciences Corporation | Uncoupling of DNA insert propagation and expression of protein for phage display |
| WO2010142785A1 (en) * | 2009-06-11 | 2010-12-16 | Institut Curie | Methods and kits for regulating intracellular trafficking of a target protein |
| US7897381B2 (en) | 2002-08-07 | 2011-03-01 | Ambit Biosciences Corporation | Uncoupling of DNA insert propagation and expression of protein for phage display |
| US8034770B2 (en) | 2008-06-04 | 2011-10-11 | Amgen Inc. | FGF21 polypeptides comprising two or more mutations |
| WO2011126808A2 (en) | 2010-03-29 | 2011-10-13 | The Trustees Of The University Of Pennsylvania | Pharmacologically induced transgene ablation system |
| US8163726B2 (en) | 2002-09-18 | 2012-04-24 | University Of Pennsylvania | Method of inhibiting choroidal neovascularization |
| US8188040B2 (en) | 2009-05-05 | 2012-05-29 | Amgen Inc. | FGF21 mutants and uses thereof |
| FR2968013A1 (en) * | 2010-11-29 | 2012-06-01 | Cis Bio Int | METHOD OF CONDITIONALLY RETAINING A PROTEIN OF INTEREST IN THE ENDOPLASMIC RETICULUM |
| WO2012145572A1 (en) | 2011-04-20 | 2012-10-26 | The Trustees Of The University Of Pennsylvania | Regimens and compositions for aav-mediated passive immunization of airborne pathogens |
| WO2013049493A1 (en) | 2011-09-28 | 2013-04-04 | The Trustees Of The University Of Pennsylvania | Inducible adeno -associated virus vector mediated transgene ablation system |
| US8426167B2 (en) | 2001-06-22 | 2013-04-23 | Roche Diagnostics Operations, Inc. | Methods for producing fusion polypeptides or enhancing expression of fusion polypeptides |
| US8486960B2 (en) | 2006-03-23 | 2013-07-16 | Santen Pharmaceutical Co., Ltd. | Formulations and methods for vascular permeability-related diseases or conditions |
| US8658667B2 (en) | 2006-02-09 | 2014-02-25 | Santen Pharmaceutical Co., Ltd. | Stable formulations, and methods of their preparation and use |
| US8927005B2 (en) | 2005-02-09 | 2015-01-06 | Santen Pharmaceutical Co., Ltd. | Liquid formulations for treatment of diseases or conditions |
| US8931331B2 (en) | 2011-05-18 | 2015-01-13 | 3M Innovative Properties Company | Systems and methods for volumetric metering on a sample processing device |
| US9067205B2 (en) | 2011-05-18 | 2015-06-30 | 3M Innovative Properties Company | Systems and methods for valving on a sample processing device |
| US9168523B2 (en) | 2011-05-18 | 2015-10-27 | 3M Innovative Properties Company | Systems and methods for detecting the presence of a selected volume of material in a sample processing device |
| US9267165B2 (en) | 2002-04-02 | 2016-02-23 | Discoverx Corporation | Assays and kits for detecting protein binding |
| US9279013B2 (en) | 2008-10-10 | 2016-03-08 | Amgen Inc. | FGF-21 mutants comprising polyethylene glycol and uses thereof |
| US9284378B2 (en) | 2009-12-07 | 2016-03-15 | Shaw-Fen Sylvia Hu | Human antigen binding proteins that bind β-Klotho, FGF receptors and complexes thereof |
| WO2016100236A3 (en) * | 2014-12-15 | 2016-09-09 | Bellicum Pharmaceuticals, Inc. | Methods for controlled elimination of therapeutic cells |
| WO2016098078A3 (en) * | 2014-12-19 | 2016-10-27 | Novartis Ag | Dimerization switches and uses thereof |
| US9493530B2 (en) | 2009-05-05 | 2016-11-15 | Amgen Inc. | FGF21 mutants comprising a mutation at position 98, 171 and/or 180 |
| WO2016196655A1 (en) | 2015-06-03 | 2016-12-08 | The Regents Of The University Of California | Cas9 variants and methods of use thereof |
| US9517264B2 (en) | 2010-04-15 | 2016-12-13 | Amgen Inc. | Human FGF receptor and β-Klotho binding proteins |
| WO2017075335A1 (en) | 2015-10-28 | 2017-05-04 | Voyager Therapeutics, Inc. | Regulatable expression using adeno-associated virus (aav) |
| WO2017087723A1 (en) | 2015-11-19 | 2017-05-26 | The Regents Of The University Of California | Conditionally repressible immune cell receptors and methods of use thereof |
| US9719106B2 (en) | 2013-04-29 | 2017-08-01 | The Trustees Of The University Of Pennsylvania | Tissue preferential codon modified expression cassettes, vectors containing same, and uses thereof |
| CN107207621A (en) * | 2015-02-24 | 2017-09-26 | Ucl商务股份有限公司 | Chimeric protein |
| EP3293257A1 (en) | 2009-03-20 | 2018-03-14 | Mesoblast, Inc. | Production of reprogrammed pluripotent cells |
| EP3300745A1 (en) | 2013-02-15 | 2018-04-04 | The Regents of the University of California | Chimeric antigen receptor and methods of use thereof |
| WO2018160573A1 (en) | 2017-02-28 | 2018-09-07 | The Trustees Of The University Of Pennsylvania | Influenza vaccines based on aav vectors |
| WO2018161064A1 (en) | 2017-03-03 | 2018-09-07 | F1 Oncology, Inc. | Methods and compositions for transducing and expanding lymphocytes and regulating the activity thereof |
| WO2019064182A1 (en) | 2017-09-26 | 2019-04-04 | Novartis Ag | Rapamycin derivatives |
| WO2019079496A2 (en) | 2017-10-18 | 2019-04-25 | Regenxbio, Inc. | Fully-human post-translationally modified antibody therapeutics |
| WO2019079494A1 (en) | 2017-10-18 | 2019-04-25 | Regenxbio, Inc. | Treatment of ocular diseases and metastatic colon cancer with human post-translationally modified vegf-trap |
| CN110191955A (en) * | 2016-12-13 | 2019-08-30 | 西雅图儿童医院(Dba西雅图儿童研究所) | The method that exogenous drugs activation is carried out to the signal transduction compound for the chemical substance induction expressed in the cell of engineering in vitro and in vivo |
| US10441644B2 (en) | 2015-05-05 | 2019-10-15 | The Regents Of The University Of California | H3.3 CTL peptides and uses thereof |
| WO2020047527A2 (en) | 2018-09-02 | 2020-03-05 | F1 Bioventures, Llc | Methods and compositions for genetically modifying lymphocytes in blood or in enriched pbmcs |
| EP3632461A1 (en) | 2018-10-05 | 2020-04-08 | St. Anna Kinderkrebsforschung | A group of chimeric antigen receptors (cars) |
| EP3632460A1 (en) | 2018-10-05 | 2020-04-08 | St. Anna Kinderkrebsforschung | A group of chimeric antigen receptors (cars) |
| WO2020070290A1 (en) | 2018-10-05 | 2020-04-09 | St. Anna Kinderkrebsforschung | A group of chimeric antigen receptors (cars) |
| WO2020070289A1 (en) | 2018-10-05 | 2020-04-09 | St. Anna Kinderkrebsforschung | A group of chimeric antigen receptors (cars) |
| EP3663405A1 (en) | 2013-06-11 | 2020-06-10 | Takara Bio USA, Inc. | Protein enriched microvesicles and methods of making and using the same |
| WO2020128861A1 (en) * | 2018-12-18 | 2020-06-25 | Novartis Ag | Rapamycin derivatives |
| WO2020206098A1 (en) | 2019-04-03 | 2020-10-08 | Regenxbio Inc. | Gene therapy for eye pathologies |
| WO2020219868A1 (en) | 2019-04-24 | 2020-10-29 | Regenxbio Inc. | Fully-human post-translationally modified antibody therapeutics |
| WO2021041373A1 (en) | 2019-08-26 | 2021-03-04 | Regenxbio Inc. | Treatment of diabetic retinopathy with fully-human post-translationally modified anti-vegf fab |
| WO2021071835A1 (en) | 2019-10-07 | 2021-04-15 | Regenxbio Inc. | Adeno-associated virus vector pharmaceutical composition and methods |
| US11197937B2 (en) | 2016-04-15 | 2021-12-14 | The Trustees Of The University Of Pennsylvania | Compositions for treatment of wet age-related macular degeneration |
| WO2022060916A1 (en) | 2020-09-15 | 2022-03-24 | Regenxbio Inc. | Vectorized antibodies for anti-viral therapy |
| WO2022060915A1 (en) | 2020-09-15 | 2022-03-24 | Regenxbio Inc. | Vectorized lanadelumab and administration thereof |
| WO2022076582A1 (en) | 2020-10-07 | 2022-04-14 | Regenxbio Inc. | Gene therapy for ocular manifestations of cln2 disease |
| WO2022094255A2 (en) | 2020-10-29 | 2022-05-05 | Regenxbio Inc. | Vectorized factor xii antibodies and administration thereof |
| WO2022094157A1 (en) | 2020-10-28 | 2022-05-05 | Regenxbio Inc. | Vectorized anti-cgrp and anti-cgrpr antibodies and administration thereof |
| WO2022094295A1 (en) | 2020-10-29 | 2022-05-05 | Regenxbio Inc. | Vectorized tnf-alpha antagonists for ocular indications |
| WO2022094106A1 (en) | 2020-10-28 | 2022-05-05 | Regenxbio, Inc. | VECTORIZED ANTI-TNF-α ANTIBODIES FOR OCULAR INDICATIONS |
| WO2022119871A2 (en) | 2020-12-01 | 2022-06-09 | The Trustees Of The University Of Pennsylvania | Novel compositions with tissue-specific targeting motifs and compositions containing same |
| WO2022187289A1 (en) | 2021-03-01 | 2022-09-09 | Exuma Biotech Corp. | Methods and compositions for the delivery of retroviral particles |
| WO2022226263A1 (en) | 2021-04-23 | 2022-10-27 | The Trustees Of The University Of Pennsylvania | Novel compositions with brain-specific targeting motifs and compositions containing same |
| US11535665B2 (en) | 2015-05-13 | 2022-12-27 | The Trustees Of The University Of Pennsylvania | AAV-mediated expression of anti-influenza antibodies and methods of use thereof |
| US11578341B2 (en) | 2017-02-28 | 2023-02-14 | The Trustees Of The University Of Pennsylvania | Compositions useful in treatment of spinal muscular atrophy |
| WO2023056399A1 (en) | 2021-10-02 | 2023-04-06 | The Trustees Of The University Of Pennsylvania | Novel aav capsids and compositions containing same |
| WO2023147304A1 (en) | 2022-01-25 | 2023-08-03 | The Trustees Of The University Of Pennsylvania | Aav capsids for improved heart transduction and detargeting of liver |
| WO2023168305A1 (en) | 2022-03-01 | 2023-09-07 | Exuma Biotech Corp. | Viral particles with membrane-bound hyaluronidase |
| WO2023201308A1 (en) | 2022-04-14 | 2023-10-19 | Regenxbio Inc. | Gene therapy for treating an ocular disease |
| WO2023205610A2 (en) | 2022-04-18 | 2023-10-26 | Regenxbio Inc. | Hybrid aav capsids |
| WO2023215806A2 (en) | 2022-05-03 | 2023-11-09 | Regenxbio Inc. | Vectorized anti-complement antibodies and complement agents and administration thereof |
| WO2023215807A1 (en) | 2022-05-03 | 2023-11-09 | Regenxbio Inc. | VECTORIZED ANTI-TNF-α INHIBITORS FOR OCULAR INDICATIONS |
| US11819476B2 (en) | 2019-12-05 | 2023-11-21 | Janssen Pharmaceutica Nv | Rapamycin analogs and uses thereof |
| US11827906B2 (en) | 2017-02-28 | 2023-11-28 | The Trustees Of The University Of Pennsylvania | Adeno-associated virus (AAV) clade f vector and uses therefor |
| US11944605B2 (en) | 2018-06-15 | 2024-04-02 | Janssen Pharmaceutica Nv | Rapamycin analogs and uses thereof |
| WO2024073669A1 (en) | 2022-09-30 | 2024-04-04 | Regenxbio Inc. | Treatment of ocular diseases with recombinant viral vectors encoding anti-vegf fab |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994018317A1 (en) * | 1993-02-12 | 1994-08-18 | The Board Of Trustees Of The Leland Stanford Junior University | Regulated transcription of targeted genes and other biological events |
| WO1995002684A1 (en) * | 1993-07-16 | 1995-01-26 | The Board Of Trustees Of The Leland Stanford Junior University | Regulated apoptosis |
| WO1995033052A1 (en) * | 1994-05-27 | 1995-12-07 | Mitotix, Inc. | Immunosuppressant target proteins |
| WO1996006111A1 (en) * | 1994-08-18 | 1996-02-29 | Ariad Gene Therapeutics, Inc. | Regulatable elimination of gene expression, gene product function and engineered host cells |
-
1996
- 1996-06-07 KR KR1019970709226A patent/KR19990022651A/en not_active Application Discontinuation
- 1996-06-07 WO PCT/US1996/009948 patent/WO1996041865A1/en not_active Application Discontinuation
- 1996-06-07 CA CA002219080A patent/CA2219080A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994018317A1 (en) * | 1993-02-12 | 1994-08-18 | The Board Of Trustees Of The Leland Stanford Junior University | Regulated transcription of targeted genes and other biological events |
| WO1995002684A1 (en) * | 1993-07-16 | 1995-01-26 | The Board Of Trustees Of The Leland Stanford Junior University | Regulated apoptosis |
| WO1995033052A1 (en) * | 1994-05-27 | 1995-12-07 | Mitotix, Inc. | Immunosuppressant target proteins |
| WO1996006111A1 (en) * | 1994-08-18 | 1996-02-29 | Ariad Gene Therapeutics, Inc. | Regulatable elimination of gene expression, gene product function and engineered host cells |
Non-Patent Citations (3)
| Title |
|---|
| J. CHEN ET AL.: "Identification of an 11-kDa FKBP 12-rapamycin-binding domain within the 289-kDa FKBP-rapamycin-associated protein and characterization of a critical serine residue", PROC. NATL.ACAD SCI., vol. 92, May 1995 (1995-05-01), NATL. ACAD SCI.,WASHINGTON,DC,US;, pages 4947 - 4051, XP002018161 * |
| J.L. POMERANTZ ET AL.: "Structure-based design of transcription factors", SCIENCE, vol. 267, 6 January 1995 (1995-01-06), AAAS,WASHINGTON,DC,US, pages 93 - 96, XP002018160 * |
| RIVERA, VICTOR M. ET AL: "A humanized system for pharmacologic control of gene expression", NAT. MED. (N. Y.) (1996), 2(9), 1028-1032 CODEN: NAMEFI;ISSN: 1078-8956, September 1996 (1996-09-01), XP002018162 * |
Cited By (187)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6270998B1 (en) | 1991-04-26 | 2001-08-07 | Osaka Bioscience Institute | DNA coding for human cell surface antigen |
| US6949360B2 (en) | 1991-04-26 | 2005-09-27 | Osaka Bioscience Institute | DNA coding for human cell surface antigen |
| US6187757B1 (en) | 1995-06-07 | 2001-02-13 | Ariad Pharmaceuticals, Inc. | Regulation of biological events using novel compounds |
| US6649595B2 (en) | 1995-06-07 | 2003-11-18 | Ariad Gene Therapeutics, Inc. | Regulation of biological events using novel compounds |
| US6506379B1 (en) | 1995-06-07 | 2003-01-14 | Ariad Gene Therapeutics, Inc. | Intramuscular delivery of recombinant AAV |
| US6306649B1 (en) | 1995-06-27 | 2001-10-23 | Ariad Gene Therapeutics, Inc. | Heterologous transcription factors |
| US6939689B2 (en) | 1995-12-07 | 2005-09-06 | Diversa Corporation | Exonuclease-mediated nucleic acid reassembly in directed evolution |
| US6713279B1 (en) | 1995-12-07 | 2004-03-30 | Diversa Corporation | Non-stochastic generation of genetic vaccines and enzymes |
| US6773900B2 (en) | 1995-12-07 | 2004-08-10 | Diversa Corporation | End selection in directed evolution |
| US6740506B2 (en) | 1995-12-07 | 2004-05-25 | Diversa Corporation | End selection in directed evolution |
| US6171820B1 (en) | 1995-12-07 | 2001-01-09 | Diversa Corporation | Saturation mutagenesis in directed evolution |
| US6696275B2 (en) | 1995-12-07 | 2004-02-24 | Diversa Corporation | End selection in directed evolution |
| US6635449B2 (en) | 1995-12-07 | 2003-10-21 | Diversa Corporation | Exonuclease-mediated nucleic acid reassembly in directed evolution |
| US6713281B2 (en) | 1995-12-07 | 2004-03-30 | Diversa Corporation | Directed evolution of thermophilic enzymes |
| US6238884B1 (en) | 1995-12-07 | 2001-05-29 | Diversa Corporation | End selection in directed evolution |
| US7018793B1 (en) | 1995-12-07 | 2006-03-28 | Diversa Corporation | Combinatorial screening of mixed populations of organisms |
| US6713282B2 (en) | 1995-12-07 | 2004-03-30 | Diversa Corporation | End selection in directed evolution |
| US6709841B2 (en) | 1995-12-07 | 2004-03-23 | Diversa Corporation | Exonuclease-mediated gene assembly in directed evolution |
| US6352842B1 (en) | 1995-12-07 | 2002-03-05 | Diversa Corporation | Exonucease-mediated gene assembly in directed evolution |
| US6358709B1 (en) | 1995-12-07 | 2002-03-19 | Diversa Corporation | End selection in directed evolution |
| US6361974B1 (en) | 1995-12-07 | 2002-03-26 | Diversa Corporation | Exonuclease-mediated nucleic acid reassembly in directed evolution |
| WO1999030164A1 (en) * | 1997-12-09 | 1999-06-17 | President And Fellows Of Harvard College | Method to identify transcriptional modulators |
| US6153383A (en) * | 1997-12-09 | 2000-11-28 | Verdine; Gregory L. | Synthetic transcriptional modulators and uses thereof |
| US6183965B1 (en) | 1997-12-09 | 2001-02-06 | President And Fellows Of Harvard College | Synthetic transcriptional modulators and uses thereof |
| WO1999036553A3 (en) * | 1998-01-15 | 1999-10-21 | Ariad Gene Therapeutics Inc | Regulation of biological events using multimeric chimeric proteins |
| AU755784B2 (en) * | 1998-01-15 | 2002-12-19 | Ariad Pharmaceuticals, Inc. | Regulation of biological events using multimeric chimeric proteins |
| WO1999036553A2 (en) * | 1998-01-15 | 1999-07-22 | Ariad Gene Therapeutics, Inc. | Regulation of biological events using multimeric chimeric proteins |
| US6984635B1 (en) | 1998-02-13 | 2006-01-10 | Board Of Trustees Of The Leland Stanford Jr. University | Dimerizing agents, their production and use |
| WO1999058700A1 (en) * | 1998-05-11 | 1999-11-18 | Ariad Gene Therapeutics, Inc. | Multiviral compositions and uses thereof |
| WO2000004178A1 (en) * | 1998-07-14 | 2000-01-27 | Aventis Pharma Deutschland Gmbh | Expression system containing chimeric promoters with binding sites for recombinant transcription factors |
| WO2000023600A1 (en) * | 1998-10-19 | 2000-04-27 | Ariad Gene Therapeutics, Inc. | Materials and methods involving conditional aggregation domains |
| US7109317B1 (en) | 1998-11-06 | 2006-09-19 | President And Fellows Of Harvard College | FK506-based regulation of biological events |
| WO2000052179A3 (en) * | 1999-03-03 | 2000-12-21 | Genelabs Tech Inc | Dna binding compound-mediated molecular switch system |
| WO2000052179A2 (en) * | 1999-03-03 | 2000-09-08 | Genelabs Technologies, Inc. | Dna binding compound-mediated molecular switch system |
| US7189506B1 (en) | 1999-03-03 | 2007-03-13 | Genelabs Technologies, Inc. | DNA binding compound-mediated molecular switch system |
| US7579180B2 (en) | 1999-03-26 | 2009-08-25 | Amgen Inc. | Beta secretase polypeptides |
| US7582465B2 (en) | 1999-03-26 | 2009-09-01 | Amgen Inc. | Beta secretase genes |
| WO2001000815A1 (en) * | 1999-05-28 | 2001-01-04 | Sangamo Biosciences, Inc. | Molecular switches |
| US6537776B1 (en) | 1999-06-14 | 2003-03-25 | Diversa Corporation | Synthetic ligation reassembly in directed evolution |
| WO2001014387A1 (en) * | 1999-08-24 | 2001-03-01 | Ariad Gene Therapeutics, Inc. | 28-epirapalogs |
| US7196192B2 (en) | 1999-08-24 | 2007-03-27 | Ariad Gene Therapeutics, Inc. | 28-epirapalogs |
| US7067526B1 (en) | 1999-08-24 | 2006-06-27 | Ariad Gene Therapeutics, Inc. | 28-epirapalogs |
| US7887799B2 (en) | 1999-09-07 | 2011-02-15 | Amgen Inc. | Antibodies to fibroblast growth factor-like polypeptides |
| US8044024B2 (en) | 1999-09-07 | 2011-10-25 | Amgen Inc. | Identifying modulators of fibroblast growth factor-like polypeptides |
| US8053408B2 (en) | 1999-09-07 | 2011-11-08 | Amgen Inc. | Methods for treating obesity using fibroblast growth factor-like polypeptides |
| US8030275B2 (en) | 1999-09-07 | 2011-10-04 | Amgen Inc. | Methods for treating obesity using fibroblast growth factor-like polypeptides |
| EP2284189A2 (en) | 1999-09-07 | 2011-02-16 | Amgen, Inc | Nucleic acid molecules encoding fibroblast growth factor-like (FGF-like) polypeptides |
| US7879323B2 (en) | 1999-09-07 | 2011-02-01 | Amgen Inc. | Antibodies to fibroblast growth factor-like polypeptides |
| EP2258722A2 (en) | 1999-09-07 | 2010-12-08 | Amgen, Inc | Antibodies to fibroblast growth factor-like (FGF-like) polypeptides |
| US7727742B2 (en) | 1999-09-07 | 2010-06-01 | Amgen Inc. | Nucleic acid molecules encoding fibroblast growth factor-like polypeptides |
| US7408047B1 (en) | 1999-09-07 | 2008-08-05 | Amgen Inc. | Fibroblast growth factor-like polypeptides |
| US7459540B1 (en) | 1999-09-07 | 2008-12-02 | Amgen Inc. | Fibroblast growth factor-like polypeptides |
| EP2189475A2 (en) | 1999-09-07 | 2010-05-26 | Amgen Inc. | Fibroblast growth factor-like polypeptides |
| US7704952B2 (en) | 1999-09-07 | 2010-04-27 | Amgen Inc. | Methods for treating diabetes using fibroblast growth factor-like polypeptides |
| US7700558B2 (en) | 1999-09-07 | 2010-04-20 | Amgen Inc. | Methods for treating diabetes using fibroblast growth factor-like polypeptides |
| US7696172B2 (en) | 1999-09-07 | 2010-04-13 | Amgen Inc. | Fibroblast growth factor-like polypeptides |
| US7695938B2 (en) | 1999-09-07 | 2010-04-13 | Amgen Inc. | Vectors and recombinant host cells comprising nucleic acid molecules encoding Fibroblast Growth Factor-like polypeptides |
| US7671180B2 (en) | 1999-09-07 | 2010-03-02 | Amgen, Inc. | Fibroblast growth factor-like polypeptides and variants thereof |
| US7667008B2 (en) | 1999-09-07 | 2010-02-23 | Amgen, Inc. | Fibroblast growth factor-like polypeptides and variants thereof |
| US6900043B1 (en) | 1999-09-21 | 2005-05-31 | Amgen Inc. | Phosphatases which activate map kinase pathways |
| US7195902B2 (en) | 1999-09-21 | 2007-03-27 | Amgen Corporation | Phosphatases which activate map kinase pathways |
| US6562594B1 (en) | 1999-09-29 | 2003-05-13 | Diversa Corporation | Saturation mutagenesis in directed evolution |
| WO2001068705A2 (en) | 2000-03-16 | 2001-09-20 | Amgen Inc. | Il-17 receptor like molecules and uses thereof |
| EP2130920A2 (en) | 2000-03-28 | 2009-12-09 | Amgen, Inc | Beta-like glycoprotein hormone polypeptide and heterodimer |
| EP2077279A1 (en) | 2000-06-28 | 2009-07-08 | Amgen Inc. | Thymic stromal lymphopoietin receptor molecules and uses thereof |
| US8426167B2 (en) | 2001-06-22 | 2013-04-23 | Roche Diagnostics Operations, Inc. | Methods for producing fusion polypeptides or enhancing expression of fusion polypeptides |
| WO2003027671A3 (en) * | 2001-09-25 | 2004-03-11 | Eirx Therapeutics Ltd | Apoptosis |
| WO2003027671A2 (en) * | 2001-09-25 | 2003-04-03 | Eirx Therapeutics Limited | Apoptosis |
| EP2407473A2 (en) | 2002-02-01 | 2012-01-18 | ARIAD Pharmaceuticals, Inc | Method for producing phosphorus-containing compounds |
| WO2003064383A2 (en) | 2002-02-01 | 2003-08-07 | Ariad Gene Therapeutics, Inc. | Phosphorus-containing compounds & uses thereof |
| US7662924B2 (en) | 2002-02-22 | 2010-02-16 | The Board Of Trustees Of The University Of Illinois | Beta chain-associated regulator of apoptosis |
| US9267165B2 (en) | 2002-04-02 | 2016-02-23 | Discoverx Corporation | Assays and kits for detecting protein binding |
| US7833741B2 (en) | 2002-08-07 | 2010-11-16 | Ambit Biosciences Corporation | Uncoupling of DNA insert propagation and expression of protein for phage display |
| US7897381B2 (en) | 2002-08-07 | 2011-03-01 | Ambit Biosciences Corporation | Uncoupling of DNA insert propagation and expression of protein for phage display |
| US8163726B2 (en) | 2002-09-18 | 2012-04-24 | University Of Pennsylvania | Method of inhibiting choroidal neovascularization |
| US8618088B2 (en) | 2002-09-18 | 2013-12-31 | University Of Pennsylvania | Methods of inhibiting choroidal neovascularization |
| EP1699415A2 (en) * | 2003-12-29 | 2006-09-13 | Ramot at Tel-Aviv University Ltd. | An assay for the detection of rapamycin and rapamycin analogs |
| EP1699415A4 (en) * | 2003-12-29 | 2008-01-09 | Univ Ramot | An assay for the detection of rapamycin and rapamycin analogs |
| WO2005078104A1 (en) | 2004-02-09 | 2005-08-25 | Synamem Corporation | Method for generating tethered proteins |
| CN103255167A (en) * | 2004-02-09 | 2013-08-21 | 西纳门公司 | Method for producing tethered proteins |
| US9381153B2 (en) | 2005-02-09 | 2016-07-05 | Santen Pharmaceutical Co., Ltd. | Liquid formulations for treatment of diseases or conditions |
| US9387165B2 (en) | 2005-02-09 | 2016-07-12 | Santen Pharmaceutical Co., Ltd. | Rapamycin formulations and methods of their use |
| US8927005B2 (en) | 2005-02-09 | 2015-01-06 | Santen Pharmaceutical Co., Ltd. | Liquid formulations for treatment of diseases or conditions |
| US7629315B2 (en) | 2005-03-09 | 2009-12-08 | University Of Pittsburgh Of The Commonwealth System Of Higher Education | Compositions for blocking the inhibitory effect of human CRP on human leptin |
| US8658667B2 (en) | 2006-02-09 | 2014-02-25 | Santen Pharmaceutical Co., Ltd. | Stable formulations, and methods of their preparation and use |
| US9452156B2 (en) | 2006-03-23 | 2016-09-27 | Santen Pharmaceutical Co., Ltd. | Formulations and methods for vascular permeability-related diseases or conditions |
| US8486960B2 (en) | 2006-03-23 | 2013-07-16 | Santen Pharmaceutical Co., Ltd. | Formulations and methods for vascular permeability-related diseases or conditions |
| US8642546B2 (en) | 2008-06-04 | 2014-02-04 | Amgen Inc. | FGF21 mutant fusion polypeptides and uses thereof |
| US11840558B2 (en) | 2008-06-04 | 2023-12-12 | Amgen Inc. | Methods of treating non-alcoholic steatohepatitis using FGF21 mutants |
| US8410051B2 (en) | 2008-06-04 | 2013-04-02 | Amgen Inc. | FGF21 mutants and uses thereof |
| US10011642B2 (en) | 2008-06-04 | 2018-07-03 | Amgen Inc. | Methods of treating of diabetes and obesity using FGF21 mutants |
| US8361963B2 (en) | 2008-06-04 | 2013-01-29 | Amgen Inc. | Uses of FGF21 polypeptides comprising two or more mutations |
| US11072640B2 (en) | 2008-06-04 | 2021-07-27 | Amgen Inc. | Methods of treating non-alcoholic steatohepatitis using FGF21 mutants |
| US8034770B2 (en) | 2008-06-04 | 2011-10-11 | Amgen Inc. | FGF21 polypeptides comprising two or more mutations |
| US9273106B2 (en) | 2008-06-04 | 2016-03-01 | Amgen Inc. | FGF mutants with reduced proteolysis and aggregation |
| US9279013B2 (en) | 2008-10-10 | 2016-03-08 | Amgen Inc. | FGF-21 mutants comprising polyethylene glycol and uses thereof |
| EP3293257A1 (en) | 2009-03-20 | 2018-03-14 | Mesoblast, Inc. | Production of reprogrammed pluripotent cells |
| US8835385B2 (en) | 2009-05-05 | 2014-09-16 | Amgen Inc. | FGF21 polypeptides comprising two or more mutations and uses thereof |
| US8618053B2 (en) | 2009-05-05 | 2013-12-31 | Amgen Inc. | FGF21 mutants multimers and uses thereof |
| US9493530B2 (en) | 2009-05-05 | 2016-11-15 | Amgen Inc. | FGF21 mutants comprising a mutation at position 98, 171 and/or 180 |
| US8188040B2 (en) | 2009-05-05 | 2012-05-29 | Amgen Inc. | FGF21 mutants and uses thereof |
| US8795985B2 (en) | 2009-05-05 | 2014-08-05 | Amgen Inc. | FGF 21 polypeptides comprising two or more mutations and uses thereof |
| WO2010142785A1 (en) * | 2009-06-11 | 2010-12-16 | Institut Curie | Methods and kits for regulating intracellular trafficking of a target protein |
| US10570205B2 (en) | 2009-12-07 | 2020-02-25 | Amgen, Inc. | Human antigen binding proteins that bind β-Klotho, FGF receptors and complexes thereof |
| US9493577B2 (en) | 2009-12-07 | 2016-11-15 | Amgen Inc. | Human antigen binding proteins that bind β-klotho, FGF receptors and complexes thereof |
| US9284378B2 (en) | 2009-12-07 | 2016-03-15 | Shaw-Fen Sylvia Hu | Human antigen binding proteins that bind β-Klotho, FGF receptors and complexes thereof |
| WO2011126808A2 (en) | 2010-03-29 | 2011-10-13 | The Trustees Of The University Of Pennsylvania | Pharmacologically induced transgene ablation system |
| US9517264B2 (en) | 2010-04-15 | 2016-12-13 | Amgen Inc. | Human FGF receptor and β-Klotho binding proteins |
| WO2012072946A1 (en) * | 2010-11-29 | 2012-06-07 | Cis Bio International | Method for conditional retention of a protein of interest in the endoplasmic reticulum |
| FR2968013A1 (en) * | 2010-11-29 | 2012-06-01 | Cis Bio Int | METHOD OF CONDITIONALLY RETAINING A PROTEIN OF INTEREST IN THE ENDOPLASMIC RETICULUM |
| US20140031418A1 (en) * | 2011-04-20 | 2014-01-30 | The Trustees Of The University Of Pennsylvania | Regimens and Compositions for AAV-Mediated Passive Immunization of Airborne Pathogens |
| WO2012145572A1 (en) | 2011-04-20 | 2012-10-26 | The Trustees Of The University Of Pennsylvania | Regimens and compositions for aav-mediated passive immunization of airborne pathogens |
| EP3699286A1 (en) | 2011-04-20 | 2020-08-26 | The Trustees of the University of Pennsylvania | Regimens and compositions for aav-mediated passive immunization of airborne pathogens |
| US9168523B2 (en) | 2011-05-18 | 2015-10-27 | 3M Innovative Properties Company | Systems and methods for detecting the presence of a selected volume of material in a sample processing device |
| US9067205B2 (en) | 2011-05-18 | 2015-06-30 | 3M Innovative Properties Company | Systems and methods for valving on a sample processing device |
| US8931331B2 (en) | 2011-05-18 | 2015-01-13 | 3M Innovative Properties Company | Systems and methods for volumetric metering on a sample processing device |
| US9725762B2 (en) | 2011-05-18 | 2017-08-08 | Diasorin S.P.A. | Systems and methods for detecting the presence of a selected volume of material in a sample processing device |
| WO2013049493A1 (en) | 2011-09-28 | 2013-04-04 | The Trustees Of The University Of Pennsylvania | Inducible adeno -associated virus vector mediated transgene ablation system |
| EP4303232A2 (en) | 2013-02-15 | 2024-01-10 | The Regents of The University of California | Chimeric antigen receptor and methods of use thereof |
| EP3613439A1 (en) | 2013-02-15 | 2020-02-26 | The Regents Of The University Of California | Chimeric antigen receptor and methods of use thereof |
| EP3300745A1 (en) | 2013-02-15 | 2018-04-04 | The Regents of the University of California | Chimeric antigen receptor and methods of use thereof |
| EP3881868A1 (en) | 2013-02-15 | 2021-09-22 | The Regents Of The University Of California | Chimeric antigen receptor and methods of use thereof |
| US9719106B2 (en) | 2013-04-29 | 2017-08-01 | The Trustees Of The University Of Pennsylvania | Tissue preferential codon modified expression cassettes, vectors containing same, and uses thereof |
| US10647998B2 (en) | 2013-04-29 | 2020-05-12 | The Trustees Of The University Of Pennsylvania | Tissue preferential codon modified expression cassettes, vectors containing same, and uses thereof |
| EP3663405A1 (en) | 2013-06-11 | 2020-06-10 | Takara Bio USA, Inc. | Protein enriched microvesicles and methods of making and using the same |
| WO2016100236A3 (en) * | 2014-12-15 | 2016-09-09 | Bellicum Pharmaceuticals, Inc. | Methods for controlled elimination of therapeutic cells |
| WO2016098078A3 (en) * | 2014-12-19 | 2016-10-27 | Novartis Ag | Dimerization switches and uses thereof |
| CN107207621B (en) * | 2015-02-24 | 2023-04-18 | 奥托路斯有限公司 | Chimeric proteins |
| RU2746755C2 (en) * | 2015-02-24 | 2021-04-20 | Отолус Лимитед | Chimeric protein |
| US10478457B2 (en) | 2015-02-24 | 2019-11-19 | Ucl Business Ltd | Chimeric protein |
| EP3197453B1 (en) * | 2015-02-24 | 2022-08-24 | Autolus Limited | Chimeric protein |
| CN107207621A (en) * | 2015-02-24 | 2017-09-26 | Ucl商务股份有限公司 | Chimeric protein |
| US11103532B2 (en) | 2015-02-24 | 2021-08-31 | Autolus Limited | Chimeric protein |
| AU2016225190B2 (en) * | 2015-02-24 | 2021-05-13 | Autolus Limited | Chimeric protein |
| EP4147696A1 (en) * | 2015-02-24 | 2023-03-15 | Autolus Limited | Chimeric protein |
| US11185577B2 (en) | 2015-05-05 | 2021-11-30 | The Regents Of The University Of California | H3.3 CTL peptides and uses thereof |
| US10441644B2 (en) | 2015-05-05 | 2019-10-15 | The Regents Of The University Of California | H3.3 CTL peptides and uses thereof |
| US11925679B2 (en) | 2015-05-05 | 2024-03-12 | The Regents Of The University Of California | H3.3 CTL peptides and uses thereof |
| US10849965B2 (en) | 2015-05-05 | 2020-12-01 | The Regents Of The University Of California | H3.3 CTL peptides and uses thereof |
| US11535665B2 (en) | 2015-05-13 | 2022-12-27 | The Trustees Of The University Of Pennsylvania | AAV-mediated expression of anti-influenza antibodies and methods of use thereof |
| WO2016196655A1 (en) | 2015-06-03 | 2016-12-08 | The Regents Of The University Of California | Cas9 variants and methods of use thereof |
| WO2017075335A1 (en) | 2015-10-28 | 2017-05-04 | Voyager Therapeutics, Inc. | Regulatable expression using adeno-associated virus (aav) |
| WO2017087723A1 (en) | 2015-11-19 | 2017-05-26 | The Regents Of The University Of California | Conditionally repressible immune cell receptors and methods of use thereof |
| US11197937B2 (en) | 2016-04-15 | 2021-12-14 | The Trustees Of The University Of Pennsylvania | Compositions for treatment of wet age-related macular degeneration |
| CN110191955A (en) * | 2016-12-13 | 2019-08-30 | 西雅图儿童医院(Dba西雅图儿童研究所) | The method that exogenous drugs activation is carried out to the signal transduction compound for the chemical substance induction expressed in the cell of engineering in vitro and in vivo |
| US11578341B2 (en) | 2017-02-28 | 2023-02-14 | The Trustees Of The University Of Pennsylvania | Compositions useful in treatment of spinal muscular atrophy |
| WO2018160573A1 (en) | 2017-02-28 | 2018-09-07 | The Trustees Of The University Of Pennsylvania | Influenza vaccines based on aav vectors |
| US10786568B2 (en) | 2017-02-28 | 2020-09-29 | The Trustees Of The University Of Pennsylvania | AAV mediated influenza vaccines |
| US11827906B2 (en) | 2017-02-28 | 2023-11-28 | The Trustees Of The University Of Pennsylvania | Adeno-associated virus (AAV) clade f vector and uses therefor |
| WO2018161064A1 (en) | 2017-03-03 | 2018-09-07 | F1 Oncology, Inc. | Methods and compositions for transducing and expanding lymphocytes and regulating the activity thereof |
| WO2019064182A1 (en) | 2017-09-26 | 2019-04-04 | Novartis Ag | Rapamycin derivatives |
| US10800793B2 (en) | 2017-09-26 | 2020-10-13 | Novartis Ag | Rapamycin derivatives |
| WO2019079496A2 (en) | 2017-10-18 | 2019-04-25 | Regenxbio, Inc. | Fully-human post-translationally modified antibody therapeutics |
| WO2019079494A1 (en) | 2017-10-18 | 2019-04-25 | Regenxbio, Inc. | Treatment of ocular diseases and metastatic colon cancer with human post-translationally modified vegf-trap |
| EP4317185A2 (en) | 2017-10-18 | 2024-02-07 | REGENXBIO Inc. | Fully-human post-translationally modified antibody therapeutics |
| US11944605B2 (en) | 2018-06-15 | 2024-04-02 | Janssen Pharmaceutica Nv | Rapamycin analogs and uses thereof |
| WO2020047527A2 (en) | 2018-09-02 | 2020-03-05 | F1 Bioventures, Llc | Methods and compositions for genetically modifying lymphocytes in blood or in enriched pbmcs |
| WO2020070290A1 (en) | 2018-10-05 | 2020-04-09 | St. Anna Kinderkrebsforschung | A group of chimeric antigen receptors (cars) |
| EP3632461A1 (en) | 2018-10-05 | 2020-04-08 | St. Anna Kinderkrebsforschung | A group of chimeric antigen receptors (cars) |
| EP3632460A1 (en) | 2018-10-05 | 2020-04-08 | St. Anna Kinderkrebsforschung | A group of chimeric antigen receptors (cars) |
| WO2020070289A1 (en) | 2018-10-05 | 2020-04-09 | St. Anna Kinderkrebsforschung | A group of chimeric antigen receptors (cars) |
| CN113260619A (en) * | 2018-12-18 | 2021-08-13 | 诺华股份有限公司 | Rapamycin derivatives |
| WO2020128861A1 (en) * | 2018-12-18 | 2020-06-25 | Novartis Ag | Rapamycin derivatives |
| AU2019409366B2 (en) * | 2018-12-18 | 2022-10-27 | Novartis Ag | Rapamycin derivatives |
| WO2020206098A1 (en) | 2019-04-03 | 2020-10-08 | Regenxbio Inc. | Gene therapy for eye pathologies |
| WO2020219868A1 (en) | 2019-04-24 | 2020-10-29 | Regenxbio Inc. | Fully-human post-translationally modified antibody therapeutics |
| WO2021041373A1 (en) | 2019-08-26 | 2021-03-04 | Regenxbio Inc. | Treatment of diabetic retinopathy with fully-human post-translationally modified anti-vegf fab |
| WO2021071835A1 (en) | 2019-10-07 | 2021-04-15 | Regenxbio Inc. | Adeno-associated virus vector pharmaceutical composition and methods |
| US11819476B2 (en) | 2019-12-05 | 2023-11-21 | Janssen Pharmaceutica Nv | Rapamycin analogs and uses thereof |
| WO2022060915A1 (en) | 2020-09-15 | 2022-03-24 | Regenxbio Inc. | Vectorized lanadelumab and administration thereof |
| WO2022060916A1 (en) | 2020-09-15 | 2022-03-24 | Regenxbio Inc. | Vectorized antibodies for anti-viral therapy |
| WO2022076582A1 (en) | 2020-10-07 | 2022-04-14 | Regenxbio Inc. | Gene therapy for ocular manifestations of cln2 disease |
| WO2022094157A1 (en) | 2020-10-28 | 2022-05-05 | Regenxbio Inc. | Vectorized anti-cgrp and anti-cgrpr antibodies and administration thereof |
| WO2022094106A1 (en) | 2020-10-28 | 2022-05-05 | Regenxbio, Inc. | VECTORIZED ANTI-TNF-α ANTIBODIES FOR OCULAR INDICATIONS |
| WO2022094255A2 (en) | 2020-10-29 | 2022-05-05 | Regenxbio Inc. | Vectorized factor xii antibodies and administration thereof |
| WO2022094295A1 (en) | 2020-10-29 | 2022-05-05 | Regenxbio Inc. | Vectorized tnf-alpha antagonists for ocular indications |
| WO2022119871A2 (en) | 2020-12-01 | 2022-06-09 | The Trustees Of The University Of Pennsylvania | Novel compositions with tissue-specific targeting motifs and compositions containing same |
| WO2022187289A1 (en) | 2021-03-01 | 2022-09-09 | Exuma Biotech Corp. | Methods and compositions for the delivery of retroviral particles |
| WO2022226263A1 (en) | 2021-04-23 | 2022-10-27 | The Trustees Of The University Of Pennsylvania | Novel compositions with brain-specific targeting motifs and compositions containing same |
| WO2023056399A1 (en) | 2021-10-02 | 2023-04-06 | The Trustees Of The University Of Pennsylvania | Novel aav capsids and compositions containing same |
| WO2023147304A1 (en) | 2022-01-25 | 2023-08-03 | The Trustees Of The University Of Pennsylvania | Aav capsids for improved heart transduction and detargeting of liver |
| WO2023168305A1 (en) | 2022-03-01 | 2023-09-07 | Exuma Biotech Corp. | Viral particles with membrane-bound hyaluronidase |
| WO2023201308A1 (en) | 2022-04-14 | 2023-10-19 | Regenxbio Inc. | Gene therapy for treating an ocular disease |
| WO2023205610A2 (en) | 2022-04-18 | 2023-10-26 | Regenxbio Inc. | Hybrid aav capsids |
| WO2023215807A1 (en) | 2022-05-03 | 2023-11-09 | Regenxbio Inc. | VECTORIZED ANTI-TNF-α INHIBITORS FOR OCULAR INDICATIONS |
| WO2023215806A2 (en) | 2022-05-03 | 2023-11-09 | Regenxbio Inc. | Vectorized anti-complement antibodies and complement agents and administration thereof |
| WO2024073669A1 (en) | 2022-09-30 | 2024-04-04 | Regenxbio Inc. | Treatment of ocular diseases with recombinant viral vectors encoding anti-vegf fab |
Also Published As
| Publication number | Publication date |
|---|---|
| KR19990022651A (en) | 1999-03-25 |
| CA2219080A1 (en) | 1996-12-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6506379B1 (en) | Intramuscular delivery of recombinant AAV | |
| US6187757B1 (en) | Regulation of biological events using novel compounds | |
| WO1996041865A1 (en) | Rapamcycin-based regulation of biological events | |
| AU755784B2 (en) | Regulation of biological events using multimeric chimeric proteins | |
| AU783158B2 (en) | 28-epirapalogs | |
| AU766513B2 (en) | Novel dimerizing agents, their production and use | |
| US5834266A (en) | Regulated apoptosis | |
| US6153383A (en) | Synthetic transcriptional modulators and uses thereof | |
| US7067526B1 (en) | 28-epirapalogs | |
| US6984635B1 (en) | Dimerizing agents, their production and use | |
| US8084596B2 (en) | Regulated apoptosis | |
| US6306649B1 (en) | Heterologous transcription factors | |
| WO1996006111A1 (en) | Regulatable elimination of gene expression, gene product function and engineered host cells | |
| AU714904C (en) | Rapamcycin-based regulation of biological events | |
| US20030206891A1 (en) | Rapamycin-based biological regulation | |
| EP0833894B1 (en) | Rapamcycin-based regulation of biological events | |
| EP1127140A2 (en) | Fk506-based regulation of biological events |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BB BG BR BY CA CH CN CZ DE DK EE ES FI GB GE HU IS JP KE KG KP KR KZ LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK TJ TM TR TT UA UG US UZ VN AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2219080 Country of ref document: CA Ref country code: CA Ref document number: 2219080 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1019970709226 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1996921491 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1996921491 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1019970709226 Country of ref document: KR |
|
| WWR | Wipo information: refused in national office |
Ref document number: 1019970709226 Country of ref document: KR |