
Genetic Diversity and Population Structure of Fusarium oxysporum f. sp. conglutinans Race 1 in Northern China Samples

Abstract
:1. Introduction
2. Results
2.1. Strains Collection, Identification and Pathogenicity, and Race Test
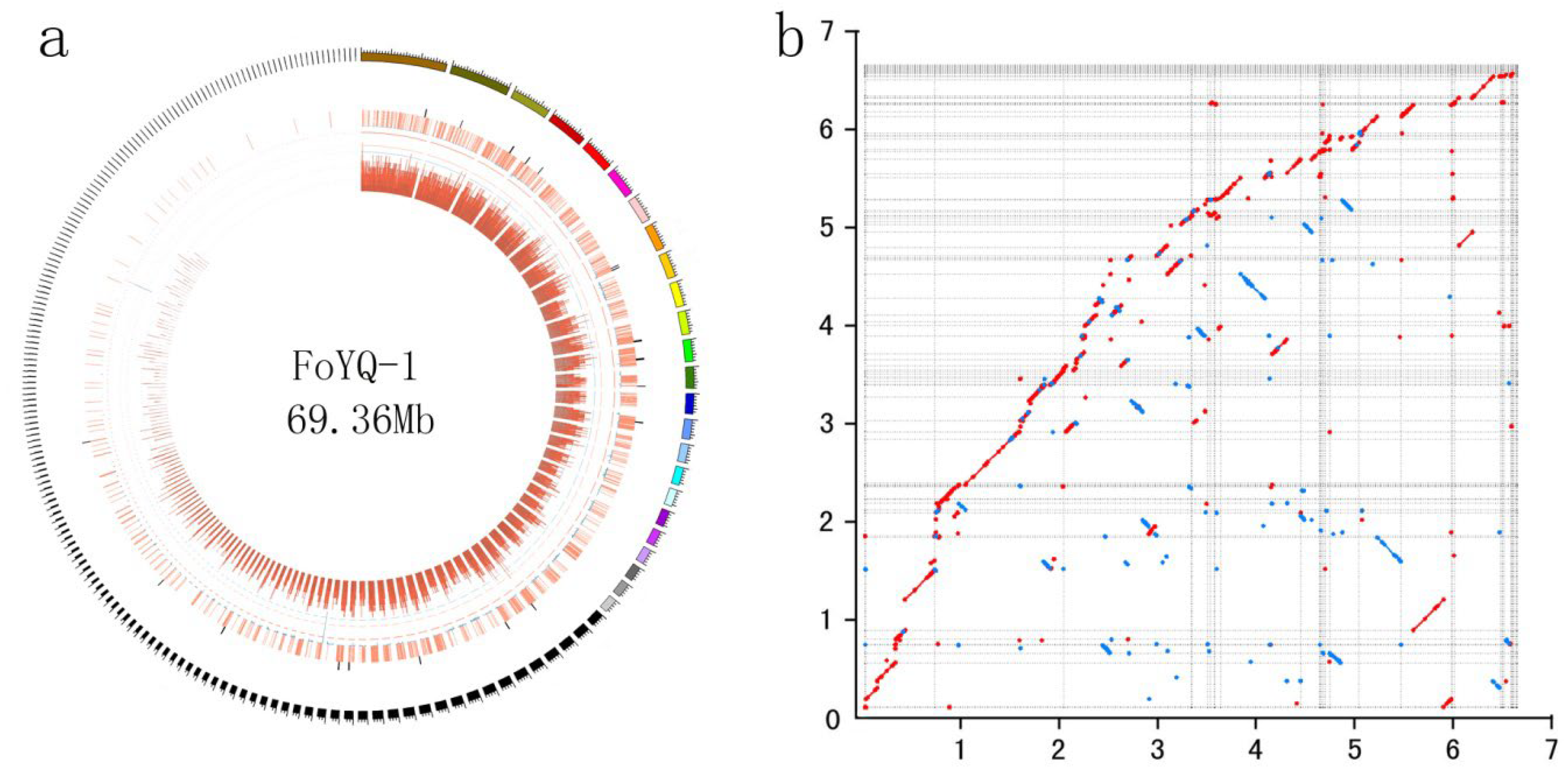
2.2. FoYQ-1 Genome Assembly and Annotation
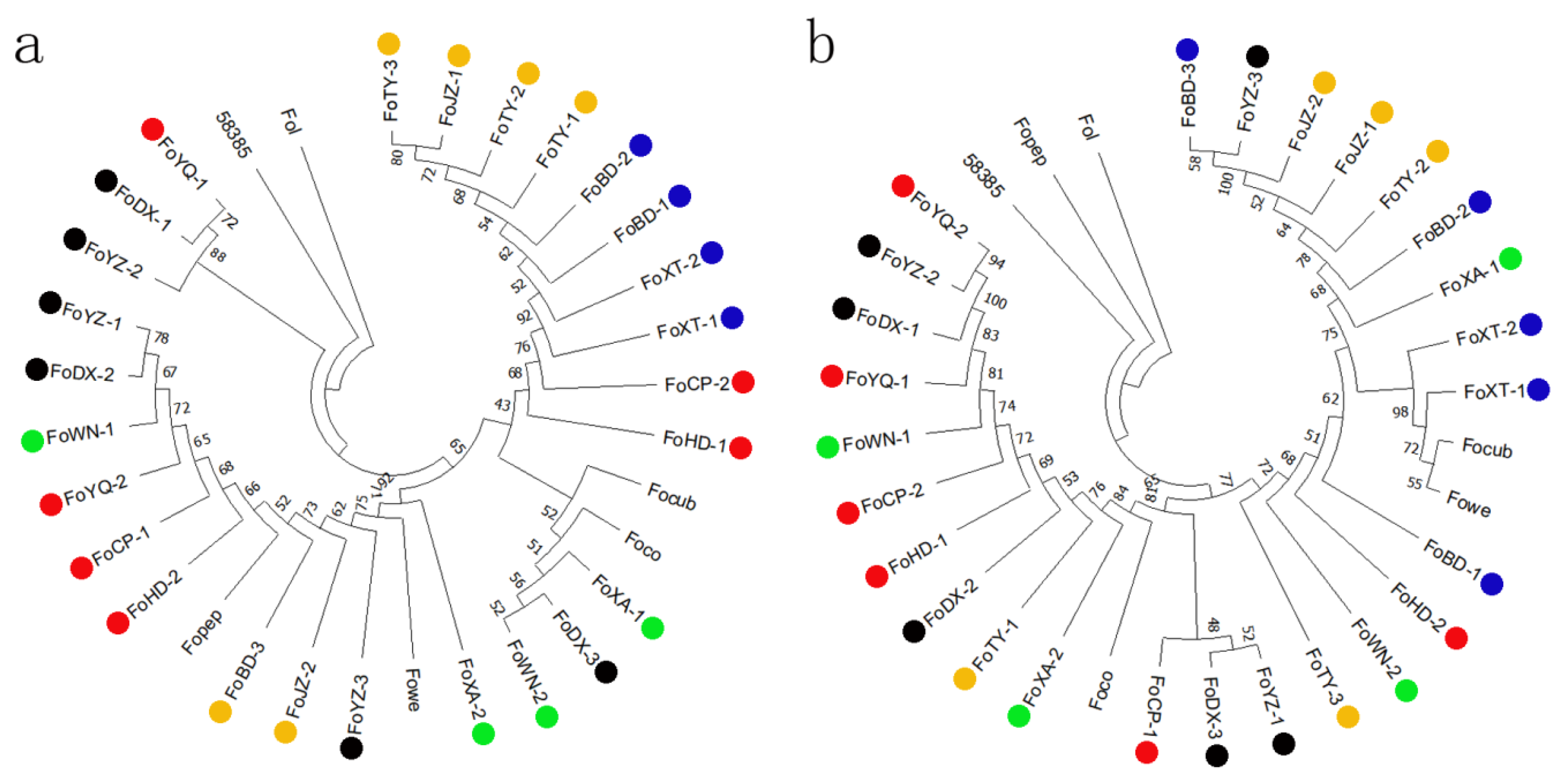
2.3. Phylogenetic Analyses and Evolutionary Relationships of FOC1
2.4. Structure Analysis of FOC1
3. Discussion
4. Methods
4.1. Isolate Collection and Race and Pathogenicity Test
4.2. Genome Assembly, Annotation, and Comparative Genomic Analysis
4.3. Molecular Characterization Based on FOC-Specific DNA Fragment and ITS and EF-1a Sequences and Construction of Phylogenetic Trees
4.4. Resequencing and SNP Calling
4.5. Diversity and Structure Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edel-Hermann, V.; Lecomte, C. Current status of Fusarium oxysporum formae speciales and races. Phytopathology 2019, 109, 512–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, P.E.; Toussoun, T.A.; Marasas, W. Fusarium Species: An Illustrated Manual for Identification; Pennsylvania State University Press: University Park, PA, USA, 1983. [Google Scholar]
- O’Donnell, K.; Ward, T.J.; Geiser, D.M.; Kistler, H.; Aoki, T. Genealogical concordance between the mating type locus and seven other nuclear genes supports formal recognition of nine phylogenetically distinct species within the Fusarium graminearum clade. Fungal Genet. Biol. 2004, 41, 600–623. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.-J.; Van Der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.-J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-Y.; Oyaizu, H.; Okada, G.; Takahashi, M. Screening of fungal antagonists against yellows of cabbage caused by Fusarium oxysporum f. sp. conglutinans. Mycoscience 2002, 43, 447–451. [Google Scholar] [CrossRef]
- Li, E.; Ling, J.; Wang, G.; Xiao, J.; Yang, Y.; Mao, Z.; Wang, X.; Xie, B. Comparative proteomics analyses of two races of Fusarium oxysporum f. sp. conglutinans that differ in pathogenicity. Sci. Rep. 2015, 5, 13663. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Villupadua, J.; Endo, R.M.; Bosland, P.; Wiliams, P.H. A new race of Fusarium oxysporum f. sp. conglutinans that attacks cabbage with type A resistance. Plant Dis. 1985, 69, 612–613. [Google Scholar] [CrossRef]
- Bosland, P.W.; Williams, P.H. An evaluation of Fusarium oxysporum from crucifers based on pathogenicity, isozyme poly-morphism, vegetative compatibility, and geographic origin. Can. J. Bot. 1987, 65, 2067–2073. [Google Scholar] [CrossRef]
- Morrison, R.H.; Mengistu, A.; Williams, P.H. First report of race 2 of cabbage yellows caused by Fusarium oxysporum f. sp. conglutinans in Texas. Plant Dis. 1994, 78, 641C. [Google Scholar] [CrossRef]
- Yang, L.; Fang, Z.; Zhuang, M.; Zhang, Y.; Lv, H.; Liu, Y. ‘12th Five-Year’ Advances in genetic breeding of cabbage in China. China Veg. 2016, 1, 1–6. [Google Scholar]
- Lv, H.; Wang, Q.B.; Yang, L.M.; Fang, Z.Y.; Liu, Y.M.; Zhuang, M.; Zhang, Y.Y.; Yang, Y.-H.; Xie, B.-Y.; Wang, X.-W. Breeding of cabbage (Brassica oleracea L. var. capitata) with fusarium wilt resistance based on microspore culture and marker-assisted selection. Euphytica 2014, 200, 465–473. [Google Scholar] [CrossRef]
- Li, M.; Zhang, T.; Li, X.; Yan, H. Fusarium wilt of cruciferae and its pathogen identification. Plant Prot. 2003, 29, 44–45. [Google Scholar]
- Lv, H.; Fang, Z.; Yang, L.; Xie, B.; Liu, Y.; Zhuang, M.; Zhang, Y.; Yang, Y. Research on screening of resistant resources to Fusarium wilt and inheritance of the resistant gene in cabbage. Acta. Hortic. Sin. 2011, 38, 875–885. [Google Scholar]
- Liu, X.; Xing, M.; Kong, C.; Fang, Z.; Yang, L.; Zhang, Y.; Wang, Y.; Ling, J.; Yang, Y.; Lv, H. Genetic Diversity, virulence, race profiling, and comparative genomic analysis of the Fusarium oxysporum f. sp. conglutinans strains infecting cabbages in China. Front. Microbiol. 2019, 10, 1373. [Google Scholar] [CrossRef]
- Halpern, H.C.; Qi, P.; Kemerait, R.C.; Brewer, M.T. Genetic diversity and population structure of races of Fusarium oxysporum causing cotton wilt. G3 Genes Genomes Genet. 2020, 10, 3261–3269. [Google Scholar] [CrossRef]
- Petkar, A.; Harris-Shultz, K.; Wang, H.; Brewer, M.T.; Sumabat, L.; Ji, P. Genetic and phenotypic diversity of Fusarium oxysporum f. sp. niveum populations from watermelon in the southeastern United States. PLoS ONE 2019, 14, e0219821. [Google Scholar] [CrossRef] [Green Version]
- Magdama, F.; Monserrate-Maggi, L.; Serrano, L.; García Onofre, J.; Jiménez-Gasco, M.D.M. Genetic Diversity of Fusarium oxysporum f. sp. cubense, the fusarium wilt pathogen of Banana, in Ecuador. Plants 2020, 9, 1133. [Google Scholar] [CrossRef]
- Wagner, T.A.; Gu, A.; Duke, S.E.; Bell, A.A.; Magill, C.; Liu, J. Genetic diversity and pathogenicity of verticillium dahliae isolates and their co-occurrence with Fusarium oxysporum f. sp. vasinfectum causing cotton wilt in xinjiang. China Plant Dis. 2021, 105, 978–985. [Google Scholar] [CrossRef]
- Bell, A.A.; Kemerait, R.C.; Ortiz, C.S.; Prom, S.; Quintana, J.; Nichols, R.L.; Liu, J. Genetic Diversity, virulence, and meloido-gyne incognita Interactions of Fusarium oxysporum isolates causing cotton wilt in Georgia. Plant Dis. 2017, 101, 948–956. [Google Scholar] [CrossRef] [Green Version]
- Stagnati, L.; Rahjoo, V.; Samayoa, L.F.; Holland, J.B.; Borrelli, V.M.G.; Busconi, M.; Lanubile, A.; Marocco, A. A Genome-Wide association study to understand the effect of Fusarium verticillioides infection on seedlings of a maize diversity panel. G3 Genes Genomes Genet. 2020, 10, 1685–1696. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, A.M.; Cabral, C.S.; Reis, A.; Fonseca, M.E.N.; Costa, H.; Ribeiro, F.H.S.; Boiteux, L.S. A three-decade survey of Brazilian Fusarium oxysporum f. sp. lycopersici races assessed by pathogenicity tests on differential tomato accessions and by molecular markers. J. Appl. Microbiol. 2021, 131, 873–884. [Google Scholar] [CrossRef]
- Leslie, J.F.; Summerell, B.A. Fusarium, laboratory workshops-a recent history. Mycotoxin Res. 2006, 22, 73–74. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, J.; Wu, X.; Shi, Y.; Gu, P.; Li, J. Investigation of occurrences and damage of cabbage wilt in Yanqing country of Beijing. Chin. Agric. Sci. Bull. 2007, 23, 315–320. [Google Scholar]
- Ling, J.; Zhang, J.-X.; Zeng, F.; Cao, Y.-X.; Xie, B.-Y.; Yang, Y.-H. Comparative genomics provide a rapid detection of Fusarium oxysporum f. sp. conglutinans. J. Integr. Agric. 2016, 15, 822–831. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yu, H.; Ma, L.-J. Accessory Chromosomes in Fusarium oxysporum. Phytopathology 2020, 110, 1488–1496. [Google Scholar] [CrossRef]
- Li, J.; Fokkens, L.; Rep, M. A single gene in Fusarium oxysporum limits host range. Mol. Plant Pathol. 2021, 22, 108–116. [Google Scholar] [CrossRef]
- Henry, P.M.; Pincot, D.D.A.; Jenner, B.N.; Borrero, C.; Aviles, M.; Nam, M.H.; Epstein, L.; Knapp, S.J.; Gordon, T.R. Horizontal chromosome transfer and independent evolution drive diversification in Fusarium oxysporum f. sp. fragariae. New Phytol. 2021, 230, 327–340. [Google Scholar] [CrossRef]
- Zaynab, M.; Fatima, M.; Abbas, S.; Sharif, Y.; Umair, M.; Zafar, M.H.; Bahadar, K. Role of secondary metabolites in plant defense against pathogens. Microb. Pathog. 2018, 124, 198–202. [Google Scholar] [CrossRef]
- Tong, L.; Zhao, C.; Liu, J.; Yang, L.; Zhuang, M.; Zhang, Y.; Wang, Y.; Ji, J.; Kuang, B.; Tang, K.; et al. Resource Screening and Inheritance Analysis of Fusarium oxysporum sp. conglutinans Race 2 Resistance in Cabbage (Brassica oleracea var. capitata). Genes 2022, 13, 1590. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.; Hill, C.M.; Wu, S.; Ruan, J.; Ma, Z.S. DBG2OLC: Efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci. Rep. 2016, 6, 31900. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, M.; Baldwin-Brown, J.G.; Long, A.D.; Emerson, J. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 2016, 44, e147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Xie, X.; Gu, X.; Zhao, J.; Ping, X.; Li, Y.; Yang, Y.; Mao, Z.; Xie, B. High-quality chromosome-level genomes of Cucumis metuliferus and Cucumis melo provide insight into Cucumis genome evolution. Plant J. 2021, 107, 136–148. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Scientific Name | Hosts | Sources | Sampling Coordinates | |
|---|---|---|---|---|---|
| 1 | FoYZ-1 | F. oxysporum f. sp. conglutinans | Cabbage | Yuzhong, Gansu | 35.9131 N/104.1622 E |
| 2 | FoYZ-2 | F. oxysporum f. sp. conglutinans | Cabbage | Yuzhong, Gansu | 35.9128 N/104.1659 E |
| 3 | FoYZ-3 | F. oxysporum f. sp. conglutinans | Cabbage | Yuzhong, Gansu | 35.9141 N/104.1573 E |
| 4 | FoYQ-1 | F. oxysporum f. sp. conglutinans | Cabbage | Yanqing, Beijing | 40.4302 N/115.9757 E |
| 5 | FoYQ-2 | F. oxysporum f. sp. conglutinans | Cabbage | Yanqing, Beijing | 40.4287 N/115.9782 E |
| 6 | FoXT-1 | F. oxysporum f. sp. conglutinans | Cabbage | Xintai, Hebei | 37.1977 N/114.6448 E |
| 7 | FoXT-2 | F. oxysporum f. sp. conglutinans | Cabbage | Xintai, Hebei | 37.1926 N/114.6465 E |
| 8 | FoXA-1 | F. oxysporum f. sp. conglutinans | Cabbage | Xi’an, Shaanxi | 34.3253 N/109.1893 E |
| 9 | FoXA-2 | F. oxysporum f. sp. conglutinans | Cabbage | Xi’an, Shaanxi | 34.3280 N/109.1932 E |
| 10 | FoWN-1 | F. oxysporum f. sp. conglutinans | Cabbage | Weinan, Shaanxi | 34.6500 N/109.6391 E |
| 11 | FoWN-2 | F. oxysporum f. sp. conglutinans | Cabbage | Weinan, Shaanxi | 34.6493 N/109.6400 E |
| 12 | FoTY-1 | F. oxysporum f. sp. conglutinans | Cabbage | Taiyuan, Shanxi | 37.9148 N/112.4041 E |
| 13 | FoTY-2 | F. oxysporum f. sp. conglutinans | Cabbage | Taiyuan, Shanxi | 37.9134 N/112.4090 E |
| 14 | FoTY-3 | F. oxysporum f. sp. conglutinans | Cabbage | Taiyuan, Shanxi | 37.9184 N/112.4021 E |
| 15 | FoJZ-1 | F. oxysporum f. sp. conglutinans | Cabbage | Jinzhong, Shanxi | 37.7784 N/112.8786 E |
| 16 | FoJZ-2 | F. oxysporum f. sp. conglutinans | Cabbage | Jinzhong, Shanxi | 37.7843 N/112.8780 E |
| 17 | FoHD-1 | F. oxysporum f. sp. conglutinans | Cabbage | Haidian, Beijing | 39.9678 N/116.3387 E |
| 18 | FoHD-2 | F. oxysporum f. sp. conglutinans | Cabbage | Haidian, Beijing | 39.9672 N/116.3398 E |
| 19 | FoDX-1 | F. oxysporum f. sp. conglutinans | Cabbage | Dingxi, Gansu | 35.5788 N/104.5490 E |
| 20 | FoDX-2 | F. oxysporum f. sp. conglutinans | Cabbage | Dingxi, Gansu | 35.5811 N/104.5496 E |
| 21 | FoDX-3 | F. oxysporum f. sp. conglutinans | Cabbage | Dingxi, Gansu | 35.5811 N/104.5496 E |
| 22 | FoCP-1 | F. oxysporum f. sp. conglutinans | Cabbage | Changping, Beijing | 40.2432 N/116.3885 E |
| 23 | FoCP-2 | F. oxysporum f. sp. conglutinans | Cabbage | Changping, Beijing | 40.2432 N/116.3851 E |
| 24 | FoBD-1 | F. oxysporum f. sp. conglutinans | Cabbage | Baoding, Hebei | 38.8809 N/115.6137 E |
| 25 | FoBD-2 | F. oxysporum f. sp. conglutinans | Cabbage | Baoding, Hebei | 38.8826 N/115.6102 E |
| 26 | FoBD-3 | F. oxysporum f. sp. conglutinans | Cabbage | Baoding, Hebei | 38.8797 N/115.6127 E |
| 27 | Fopep | F. oxysporum f. sp. capsicum | Pepper | Fujian, China | - |
| 28 | Foco | F. oxysporum f. sp. cowpea | Cowpea | Fujian, China | - |
| 29 | Focub | F. oxysporum f. sp. cucumerinum | Cucumber | Beijing, China | - |
| 30 | Fowe | F. oxysporum f. sp. melonis | Muskmelon | Fujian, China | - |
| 31 | Fol | F. oxysporum f. sp. lycopersici | Tomato | Zhejiang, China | - |
| 32 | 58,385 | F. oxysporum f. sp. conglutinans | Cabbage | ATCC | - |
| 33 | 52,557 | F. oxysporum f. sp. conglutinans | Cabbage | ATCC | - |
| Indicators of Genome Assembly | FoYQ-1 |
|---|---|
| Length of genome assembly (Mb) | 69.36 |
| Number of contigs | 192 |
| N50 of contigs (Mb) | 1.18 |
| Total length of retrotransposons (Mb) | 34.41 |
| Number of annotated genes | 18,319 |
| Average gen length (bp) | 2646 |
| Average CDS length (bp) | 1368 |
| Average protein length (bp) | 455 |
| Genome completeness (BUSCO) | 92.05% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ling, J.; Dong, X.; Ping, X.; Li, Y.; Yang, Y.; Zhao, J.; Lu, X.; Xie, B.; Mao, Z. Genetic Diversity and Population Structure of Fusarium oxysporum f. sp. conglutinans Race 1 in Northern China Samples. J. Fungi 2022, 8, 1089. https://doi.org/10.3390/jof8101089
Ling J, Dong X, Ping X, Li Y, Yang Y, Zhao J, Lu X, Xie B, Mao Z. Genetic Diversity and Population Structure of Fusarium oxysporum f. sp. conglutinans Race 1 in Northern China Samples. Journal of Fungi. 2022; 8(10):1089. https://doi.org/10.3390/jof8101089
Chicago/Turabian StyleLing, Jian, Xin Dong, Xingxing Ping, Yan Li, Yuhong Yang, Jianlong Zhao, Xiaofei Lu, Bingyan Xie, and Zhenchuan Mao. 2022. "Genetic Diversity and Population Structure of Fusarium oxysporum f. sp. conglutinans Race 1 in Northern China Samples" Journal of Fungi 8, no. 10: 1089. https://doi.org/10.3390/jof8101089